Research article
The effects of ursolic acid on cytokine production via the MAPK pathways in leukemic T-cells
Narawan Kaewthawee1, Sirikalaya Brimson2[*]
1Graduate Program in Clinical Hematology Sciences, Department of Clinical Microscopy, Faculty of Allied Health Sciences, Chulalongkorn University, Bangkok 10330, Thailand2Center for Research and Development in Molecular Hematology Sciences, Department of Clinical Microscopy, Faculty of Allied Health Sciences, Chulalongkorn University, Bangkok 10330, Thailand
EXCLI J 2013;12:Doc102
Abstract
Ursolic acid (UA) is a pentacyclic triterpenoid carboxylic acid that is found in plants and herbal products. It is one of the chemopreventive agents, which can suppress cancer cell proliferation and induce apoptosis. UA possesses various biological activities including anticancer, anti-inflammatory, anti-oxidative and hepatoprotective activity. We investigated the effect of UA on cytokine production via the MAPK pathways in Jurkat leukemic T-cells, showing that UA inhibited cell growth and proliferation of Jurkat cells, as well as suppressing PMA/PHA induced IL-2 and TNF-α production in a concentration and time dependent manner. The inhibition of IL-2 and TNF-α production by UA involved the c-Jun N-terminal kinase (JNK) pathway, but not the extracellular-signal regulated protein kinase (ERK) pathway. Future utilization of UA as a chemopreventive or therapeutic agent may provide an alternative option for leukemia treatments.
Keywords: ursolic acid, IL-2, TNF-alpha, ERK, JNK, Jurkat cells
Introduction
Leukemia is a malignancy of the blood resulting in an increase of immature white blood cells with abnormal functions. The conventional treatment for patients suffering from leukemia is chemotherapy, in which patients encounter many adverse side effects. To reduce these side effects, alternative medicines in combination with conventional treatments could provide other options for treatment of leukemia. Ursolic acid, a pentacyclic triterpenoid carboxylic acid found in plants, has various biological properties, including anti-inflammatory, anticancer, anti-angiogenic, and anti-oxidative activities (Cardenas et al., 2004[5]; Ikeda et al., 2008[15]; Manu and Kuttan, 2008[22]; Tang et al., 2009[28]; Zhang et al., 2009[32]; Wu et al., 2010[30]; Limami et al., 2011[20]; Zheng et al., 2012[33]). Apoptosis induced by UA occurs through multiple pathways, such as the inhibition of DNA replication, down-regulation of anti-apoptotic proteins, suppression of nuclear factor (NF)-κB activation, as well as other pathways (Cardenas et al., 2004[5]; Ikeda et al., 2008[14]; Manu and Kuttan, 2008[22]; Tang et al., 2009[28]; Zhang et al., 2009[32]; Yadav et al., 2010[31]). UA can inhibit gene expression of pro-inflammatory cytokines and suppress transcription factors leading to apoptosis in B16F-10 melanoma cells (Manu and Kuttan, 2008[22]). Cytokines are involved in signal transduction resulting in the activation of genes for growth and differentiation, and various other cellular activates. Pro-inflammatory cytokines such as interleukin-2 (IL-2) and tumor necrosis factor alpha (TNF-α) have both; immunostimulatory and immunosuppressive functions (O'Shea et al., 2002[25]; Bachmann and Oxenius, 2007[3]). Maximal IL-2 production in Jurkat cells can be stimulated via two signaling pathways; the first can be initiated using the ligand for the T-cell receptor (TCR) along with a CD3 specific antibody, and the second by phorbol esters (TPA) and calcium ionophore or co-stimulation (Abraham and Weiss, 2004[1]). T-cell stimulation through protein kinase Cθ (PKC) activates multiple pathways resulting in the production of many transcription factors and cytokines including IL-2 (Altman et al., 2000[2]). A previous study has shown that UA regulation of inflammatory cytokines in T- and B-lymphocytes, involves transcription factors such as NF-κB, nuclear factor of activated T cells (NF-AT) and AP-1 along with the mitogen-activated protein kinase (MAPK) pathways (Checker et al., 2012[7]).
The MAPK signaling pathways, including the signaling proteins, extracellular-signal regulated protein (ERK), c-Jun N-terminal kinase (JNK) and p38 MAPK are involved in the activation of T-cells in the immune response, regulation of gene expression, cell proliferation and programmed cell death in mammalian cells (Chang and Karin, 2001[6]; Dong et al., 2002[11]; Platanias, 2003[26]). The ERK pathway can mediate cell proliferation and apoptosis depending on the stimuli and cell type (Mebratu and Tesfaigzi, 2009[23]), whereas activation of JNK regulates T-cell differentiation in the immune system through TCR and the co-stimulation molecule CD28, resulting in the expression of IL-2 (Karin and Gallagher, 2005[17]). Other studies show that fibroblasts, which lack expression of all isoforms of JNK (JNK-null), also show severe defects in c-Jun expression, TNF-stimulated AP-1 expression and AP-1 DNA-binding activity, which are essential for cytokine production (Cloutier et al., 2003[9]; Kennedy and Davis, 2003[18]; Ventura et al., 2003[29]). JNK is also involved in both the extrinsic and intrinsic apoptotic pathways (Dhanasekaran and Reddy, 2008[10]). Furthermore, UA induced apoptosis in DU145 prostate cancer cells and human leukemia K562 cells, which occurs through the activation of the JNK pathway (Liu and Jiang, 2007[21]; Zhang et al., 2009[32]). However, the mechanism by which UA effects cytokine production in leukemia T-cells has not been fully identified. In the present study we investigated the effects of UA on IL-2 and TNF-α production via the MAPK pathways in human leukemic Jurkat T-cells.
Materials and Methods
Cell culture
Jurkat leukemic T-cell lines were cultured in RPMI-1640 (Hyclone, USA) supplemented with 10 % heat-inactivated fetal bovine serum (FBS). The cells were maintained in logarithmic growth at 37 °C in a humidified atmosphere containing a mixture of air and 5 % CO2. Ursolic acid (UA: 3β-hydroxy-12-urs-12-en-28-oic acid) (Sigma Aldrich, Switzerland) was dissolved in DMSO at 25 mM and stored at 4 °C.
Cell viability assay
Cell viability was determined using the XTT assay kit (Sigma Aldrich, USA). Jurkat cells (1x106 cells ml-1) were seeded in 96-well plates, treated with UA and maintained in normal culture conditions for 12 hours. As a control, DMSO was added to the culture at < 0.1 %. XTT working solution (20 µl) was then added into each well and incubated for 4 hours before the absorbance was read at a wavelength of 450 nm with a reference 690 nm using a microtiter plate reader (BioTek, USA). Three independent XTT experiments were performed and the final percent cell viability was calculated.
Enzyme-linked immunosorbent assay (ELISA) of IL-2 and TNF-α
Jurkat cells (1x106 cells ml-1) were treated with 20 M UA or, as a positive control, 12.5 ng ml-1 Phorbol 12-Myristate 13-Acetate (PMA) plus 0.25 µg ml-1 Lectin from Phaseolus vulgaris/Leucoagglutinin (PHA-L) (Sigma Aldrich, USA), and incubated for 1 to 24 hours. Cells were also treated with < 0.1 % DMSO as a vehicle control. To test cytokine inhibition, cells were treated with UA, 20 µM ERK inhibitor (PD98059) or 20 µM JNK inhibitor (SP600125) (Sigma Aldrich, USA) for between 0 and 12 hours prior to the addition of PMA/PHA. The cells were then incubated a further 12 hours before the culture media was collected and stored at -70 °C such that the IL-2 and TNF-α levels could be determined using the ELISA kit (R&D Systems, USA) at a later date. The absorbance was read at wavelength of 450 nm with a reference 540 nm using a microtiter plate reader (Perkin Elmer, Finland). Cytokine level was normalized to the total protein concentration of the viable cell pellets using the Bradford method with Bio-Rad protein assay reagent (Bio-Rad, CA). ELISA data were determined from three independent experiments.
Western blot analysis
Jurkat cells (1x106 cells ml-1) were treated with 20 µM UA or PMA/PHA (positive control) for between 1 and 24 hours or cells were treated with 20 µM UA for between 0 and 12 hours prior to the addition of PMA/PHA for a further 12 hours. The cells were then washed with ice-cold phosphate-buffered-saline (PBS, pH 7.4) and centrifuged at 1,500 rpm for 5 minutes three times. Whole cell lysate was prepared using lysis buffer containing 25 mM Tris-phosphate (pH 7.8) 2 mM DTT, 2mM 1,2-diaminocyclohexane-N,N,N',N'-tetraacetic acid, 10 % glycerol, 1 % Triton® X-100 and protease inhibitor cocktail (Sigma Aldrich, USA). A total of 5 µg protein extract was separated by electrophoresis on a 10 % SDS-Polyacrylamide gel and transferred onto a Polyvinylidene difluoride (PVDF) membrane (Millipore, USA). Membranes were blocked with 5 % non-fat milk in 0.1 % Tween-20 in TBS buffer for 1 hour at room temperature. The membranes were then incubated with primary antibody against p-ERK1/2, ERK1/2, SAPK/JNK, or β-actin with a dilution factor of 1:4000 or against p-SAPK/JNK with a dilution factor of 1:1000, (Cell signaling, USA) at 4 °C overnight. Membranes were then washed with 0.1 % Tween-20 in TBS buffer three times and then incubated with horseradish peroxidase (HRP) conjugated secondary antibodies (1:4000, Cell signaling, USA) for 45 minutes. Membranes were again washed three times and protein expression was detected with enhanced chemiluminescence (ECL) western blotting substrate (Pierce, USA) and to Hyperfilm-ECL (GE healthcare, UK). The intensity of the immunoreactive bands from Western blot images was analyzed using NIH Image J computer software. Western blot analyses were performed from three independent experiments.
Statistical analysis
Statistical analysis of data was performed using one-way analysis of variance (ANOVA). Data are expressed as mean values SEM. Values of P < 0.05 are considered statistically significant.
Results
Inhibition of leukemic T-cell proliferation by ursolic acid
In this study, we examined whether UA inhibited the growth of Jurkat cells in a concentration dependent manner. Cells were treated with UA (5 to 100 µM) or DMSO for 12 hours as described above and cell viability was determined using the XTT assay. The results show that UA at 30 µM and above significantly inhibits cell growth (Figure 1(Fig. 1)), giving an IC50 value of approximately 32.5 µM.
Effect of ursolic acid on IL-2 and TNF-α production
We investigated the effect of UA on cytokine production in Jurkat cells, starting with either UA or DMSO alone using ELISA, which resulted in no significant effect on IL-2 and TNF-α production (data not shown). We then treated cells with PMA/PHA (12.5 ng ml-1 / 0.25 µg ml-1) to observe the time and level of cytokine secretion. The results show that PMA/PHA stimulates IL-2 and TNF-α production significantly in a time dependent manner compared with control (DMSO) (Figure 2(Fig. 2)). IL-2 and TNF-α secretion were significantly increased at 12 hours and 3 hours respectively.
Effect of ursolic acid on inhibition of PMA/PHA-induced IL-2 and TNF-α production
Since UA failed to induce cytokine production in Jurkat cells, we therefore investigated whether UA was able to inhibit PMA/PHA-induced IL-2 and TNF-α production. The cells were pretreated with various concentrations of UA, below the IC50 (32.5 µM), and at various time points prior to the addition of PMA/PHA. Using ELISA we found that, UA significantly decreased the cytokine production compared with that of PMA/PHA alone. Furthermore, we found that UA inhibits the PMA/PHA stimulated production of IL-2 and TNF-α in a time and dose dependent manner (Figure 3a, b(Fig. 3)). Reports have previously shown that MAPK pathways are involved in cytokine production (Dong et al., 2002[11]; Platanias, 2003[26]), therefore in order to investigate whether MAPK inhibitors are sufficient to inhibit the production of IL-2 and TNF-α, we measured the level of IL-2 and TNF-α released from cells treated with PD98059 or SP600125. In contrast to UA, both inhibitors inhibit the PMA/PHA-induced IL-2 and TNF-α production without pretreatment (Figure 3c, d(Fig. 3)). However, inhibition of TNF-α production by PD98059 is less effective compared with that of SP600125. The IL-2 and TNF-α level after treatment with PMA/PHA were measured as 9,551.48 935.90 pg mg-1 protein and 938.04 85.03 pg mg-1 protein, respectively (dashed lines).
Effect of UA and PMA/PHA on MAPK activation and expression in Jurkat leukemic T-cells
We have shown in our previous results that PMA/PHA and not UA induce cytokine release from the Jurkat cells. As MAPK pathway proteins are involved in cytokine production, we therefore investigated the expression and phosphorylation of ERK and JNK proteins in cells treated with PMA/PHA or UA. Western blot analyses were performed from three independent experiments to determine the level of each protein, corresponding to the intensity of the protein bands. Western blot analysis of the proteins from cells treated with PMA/PHA (Figure 4a(Fig. 4)) represents the expression of phosphorylated ERK1/2 (p-ERK1/2), ERK1/2 and β-actin. The highest relative level of p-ERK1/2 is seen at 24 hours (Figure 4b(Fig. 4)) and relative level of ERK1/2 expression does not change compared with control cells (Figure 4c(Fig. 4)). The relative ratio between p-ERK1/2 and ERK1/2 level reveals that PMA/PHA induces maximal phosphorylation of ERK significantly at 24 hours (Figure 4d(Fig. 4)). Figure 4e(Fig. 4) represents the expression of phosphorylated JNK (p-JNK), JNK, and β-actin from cells treated with PMA/PHA. The highest significant relative level of p-JNK occurs at 1 hour but the p-JNK level is decreased significantly from 6 hours until 24 hours of treatment (Figure 4f(Fig. 4)). The relative level of JNK does not change compared with that of control cells (Figure 4g(Fig. 4)). Relative ratio between p-JNK and JNK level indicates that PMA/PHA significantly induces maximal phosphorylation of JNK at 1 hour. However, it also significantly inhibits phosphorylation of JNK from 6 hours to 24 hours (Figure 4h(Fig. 4)).
Figure 5a(Fig. 5) shows expression of p-ERK1/2, ERK1/2 and β-actin. Maximum relative level of p-ERK is found at 24 hours (Figure 5b(Fig. 5)) and there is no change in the expression level of ERK1/2 (Figure 5c(Fig. 5)) compared with that of the control. Relative ratio between p-ERK1/2 and ERK1/2 level indicates that UA, similar to PMA/PHA, significantly induces ERK phosphorylation at 24 hours (Figure 5d(Fig. 5)). Figure 5e(Fig. 5) represents expression of p-JNK, JNK and β-actin. The relative level of p-JNK is significantly reduced from 3 to 24 hours, except at 12 hours (Figure 5f(Fig. 5)), whereas relative level of JNK is decreased from 3 hours with significant reduction at 18 and 24 hours (Figure 5g(Fig. 5)). However, relative ratio between p-JNK and JNK level shows no significant difference from that of the control, which indicates that the reduction of p-JNK is not caused by the inhibition effect but rather suppression of JNK expression by UA (Figure 5h(Fig. 5)).
Inhibition of PMA/PHA-induced IL-2 and TNF-α production by ursolic acid via the JNK pathway
We have shown that PD98059, SP600125 and UA inhibited PMA/PHA-induced IL-2 and TNF-α production of Jurkat cells. However, UA inhibits IL-2 and TNF-α production in a concentration and time dependent manner, furthermore, with UA pretreatment is required. We suspected that inhibition of IL-2 and TNF-α production might involve the MAPK pathways. Therefore, at the time points where we saw inhibition of IL-2 and TNF-α production by UA, we investigated the activation and expression of MAPK proteins inside the cells. Three independent Western blot analyses were performed to determine the level of each protein. Western blot analysis shows the highest intensity of p-ERK1/2 (Figure 6a(Fig. 6)) and the maximum relative level of p-ERK1/2 at 12 hours of UA treatment prior to the addition of PMA/PHA (Figure 6b(Fig. 6)). The relative level of ERK1/2 does not change compared with that of control (Figure 6c(Fig. 6)). The relative ratio between p-ERK1/2 and ERK1/2 level shows that pre-incubation of Jurkat cells with UA 20 µM for 12 hours prior to the addition of PMA/PHA, results in maximal p-ERK1/2 level (Figure 6d(Fig. 6)). The induction of p-ERK1/2 follows a trend similar to that of cells treated with either PMA/PHA or UA alone (Figure 4d(Fig. 4), 5d(Fig. 5)).
Western blot analysis shows that a longer UA treatment time prior to the addition of PMA/PHA, results in lower expression of p-JNK and JNK (Figure 6e(Fig. 6)). The relative level of p-JNK and JNK expression shows significant reduction from 4 hours for the p-JNK and 6 hours for the JNK respectively (Figure 6f(Fig. 6) and 6g(Fig. 6)). The relative ratio between p-JNK and JNK level shows no significant difference between control and UA treatment prior to PMA/PHA stimulation (Figure 6h(Fig. 6)). This indicates that the reduction of p-JNK is caused by suppression JNK protein such that only a low amount of JNK is available for phosphorylation. From our previous results, we have shown that PMA/PHA can induce phosphorylation of JNK in the first hour of treatment. However, this result shows that pretreatment of UA can decrease PMA/ PHA-induced p-JNK due to JNK suppression.
Discussion
With anticancer properties via inhibition of cell proliferation, UA inhibits the growth of many cancerous cells such as human HT-29 colon cancer cells, Burkitt lymphoma P3HR1 cells and chronic myelogenous leukemia K562 cells with IC50 values ranging from 5-40 µM, depending on cell type and length of treatment (Chiang et al., 2003[8]; Shan et al., 2009[27]). Our study shows that UA inhibits the growth of Jurkat cells with an IC50 of 32.5 µM, percentage cell viability inversely corresponds with UA concentration and treatment time, and cell viability is significantly decreased with concentrations higher than the IC50 for UA. We also demonstrate that UA does not stimulate, rather it suppresses PMA/PHA induced IL-2 and TNF-α production in a concentration and time dependent manner. With a high concentration of UA, both IL-2 and TNF-α production are significantly inhibited at early time points. However, even at high concentrations, UA shows little inhibition of cytokine production unless it was pre-incubated with cells prior to PMA/PHA stimulation, suggesting that UA has a delayed effect on inhibition of IL-2 and TNF-α production. IL-2 is known to promote cell proliferation, inhibit apoptosis and induce cytokine production in many cells; it is also necessary for the regulation of T-cell function. TNF-α is a pro-inflammatory cytokine, which is important in the regulation of immune cells, induction of apoptosis and the induction of other cytokines (O'Shea et al., 2002[25]; Nelson, 2004[24]; Bradley, 2008[4]). Taken together, Jurkat cell proliferation is inhibited by a reduction in cytokine production. UA does not always inhibit cytokine production; according to a study showing that aggregated UA binding with CD36 induced IL-1β secretion through the p38 MAPK, ERK1/2 pathways and caspase-1 activation in RAW264.7 mouse macrophage cells (Ikeda et al., 2007[14]).
Although many studies have shown that UA can inhibit cytokines (Ikeda et al., 2008[15]; Manu and Kuttan, 2008[22]; Checker et al., 2012[7]), and induce apoptosis through JNK pathway stimulation in Jurkat cells (Liu and Jiang, 2007[21]; Dhanasekaran and Reddy, 2008[10]; Gao et al., 2012[13]), these studies have not clarified the involvement or relationship between inhibition of PMA/ PHA-induced cytokine production by UA and the MAPK signaling pathway in human leukemia T-cells. Firstly we demonstrated that IL-2 and TNF-α production are involved both the ERK and JNK pathway using PD98059 and SP600125. We also found that both UA and PD98059 inhibit IL-2 and TNF-α production but UA does not inhibit phosphorylation of ERK as PD98059 does, instead it induces phosphorylation of ERK. The results suggest that inhibition of IL-2 and TNF-α production by UA may not be related to the ERK pathway and the mechanisms of cytokine inhibition by UA and ERK inhibitor are likely to be regulated differently. Activation of the JNK pathway is required for the gene expression of many cytokines, especially IL-2 in T cells (Isakov and Altman, 2002[16]). We found that JNK expression is significantly suppressed, suggesting that the inhibition of cytokine production by UA involves the JNK pathway but not the ERK pathway. From our results, PMA/PHA induces JNK phosphorylation in the early hours of treatment and cytokine production occurs later on. UA, with pretreatment prior to PMA/PHA stimulation, does not directly inhibit PMA/PHA-induced JNK phosphorylation, but rather suppresses JNK expression. Only a low amount of JNK remains to be phosphorylated as a result of PMA/PHA treatment. Longer UA pretreatment, also significantly inhibits PMH/PHA-induced cytokine production. Taken together, JNK suppression by UA, results in insufficient JNK to be phosphorylated by PMA/PHA such that production of cytokines is consequently inhibited. Inhibition of cytokine production through JNK pathway, especially IL-2 and TNF-α in T cells leads to retardation of cell growth and activation.
However, IL-2 expression may not be restricted to the JNK pathway, as a previous study has demonstrated that expression of a dominant negative ERK mutant in Jurkat cells results in the suppression of IL-2 production (Li et al., 1999[19]). Investigations into other signaling pathway proteins and their mechanisms such as PKC, MKK4, MKK7, c-Jun or transcription factors such as AP-1 should be carried out, as they may be involved in cytokine inhibition by UA. PMA-induced PKC activation stimulates AP-1 transcription factor via the JNK pathway, resulting in an increase of IL-2 gene expression (Isakov and Altman, 2002[16]; Gaffen and Liu, 2004[12]; Karin and Gallagher, 2005[17]). However, induction of IL-2 gene expression by activation of AP-1 through the ERK pathway does not involve PKC (Gaffen and Liu, 2004[12]). Although further evaluation of the mechanisms involved is required, UA's ability to induce apoptosis and inhibit cell proliferation makes it a great candidate for use as an anticancer/leukemia drug. However, it is also necessary to investigate the toxicity of UA on normal blood cells before further clinical research into the therapeutic potential of UA.
Acknowledgements
This research was supported, in part, by the 90th Anniversary of Chulalongkorn University Fund (Ratchadaphiseksomphot Endowment Fund) and Center for Research and Development in Molecular Hematology Sciences in the Chulalongkorn University Centenary Academic Development Project. Narawan Kaewthawee received tuition fee scholarship and teaching assistant fellowship from the Department of Clinical Microscopy, Faculty of Allied Health Sciences, Chulalongkorn University. We thank Dr. James M. Brimson for advice in our research and Christopher A. Brimson for help drafting our manuscript.
References
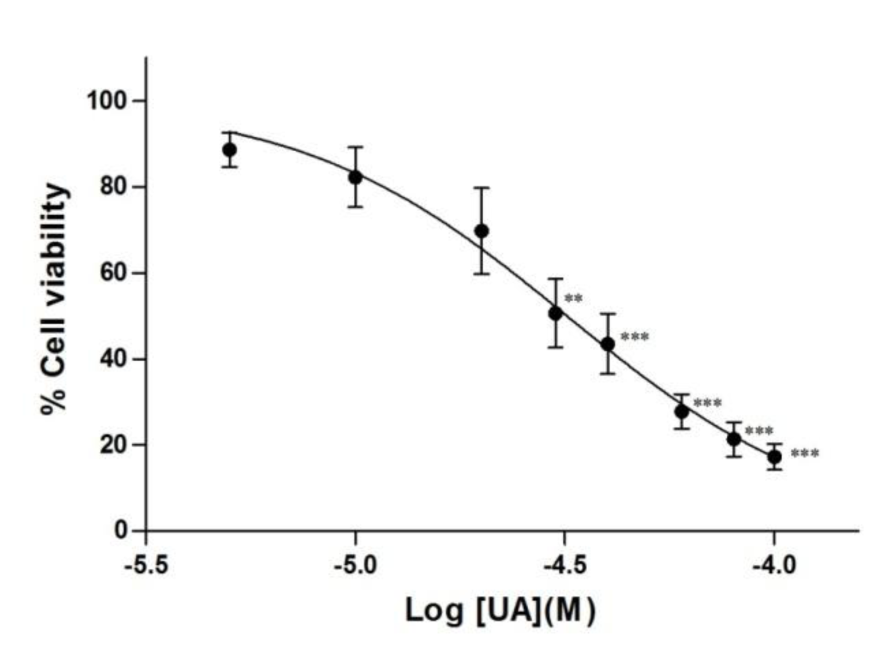
Figure 1: Inhibition of leukemic T-cell growth by ursolic acid. Jurkat cells were treated with UA (5 to 100 M) or DMSO for 12 hours. Cell viability was determined using the XTT assay. Results are expressed as mean SEM from three independent experiments. Statistical analysis was carried out with one-way ANOVA followed by Tukey's multiple comparison test compared with DMSO (** P < 0.01, *** P < 0.001).
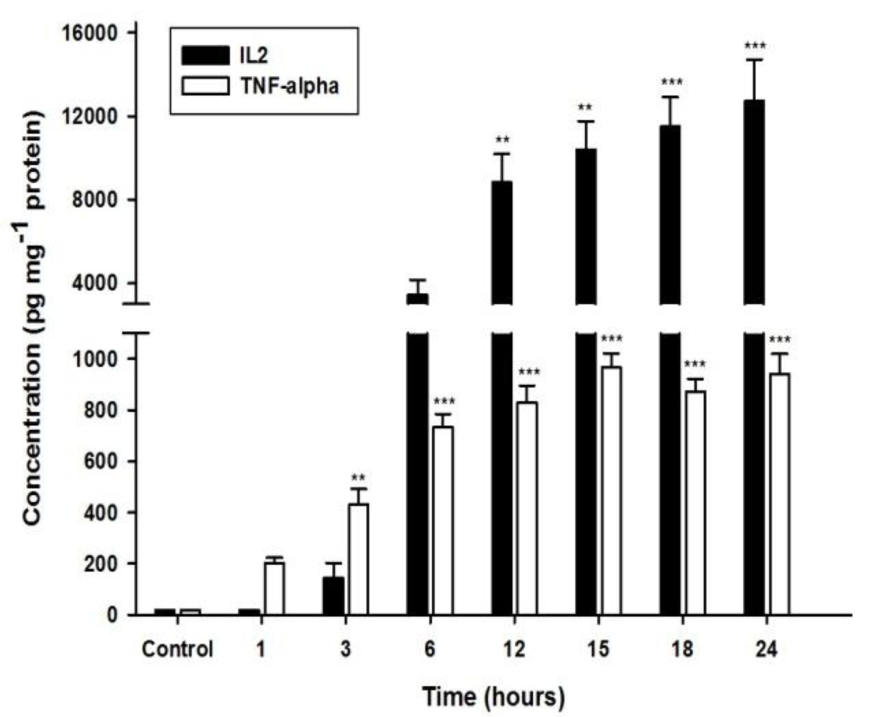
Figure 2: Effect of PMA/PHA on IL-2 and TNF-α production in Jurkat cells. Cells were treated with PMA/PHA (12.5 ng ml-1 / 0.25 µg ml-1) for 1 to 24 hours. IL-2 and TNF-α released to the media were determined using ELISA. Results are expressed as mean SEM from three independent experiments. Statistical analysis was carried out with one-way ANOVA followed by Tukey's multiple comparison test compared with DMSO control (** P < 0.01, *** P < 0.001).
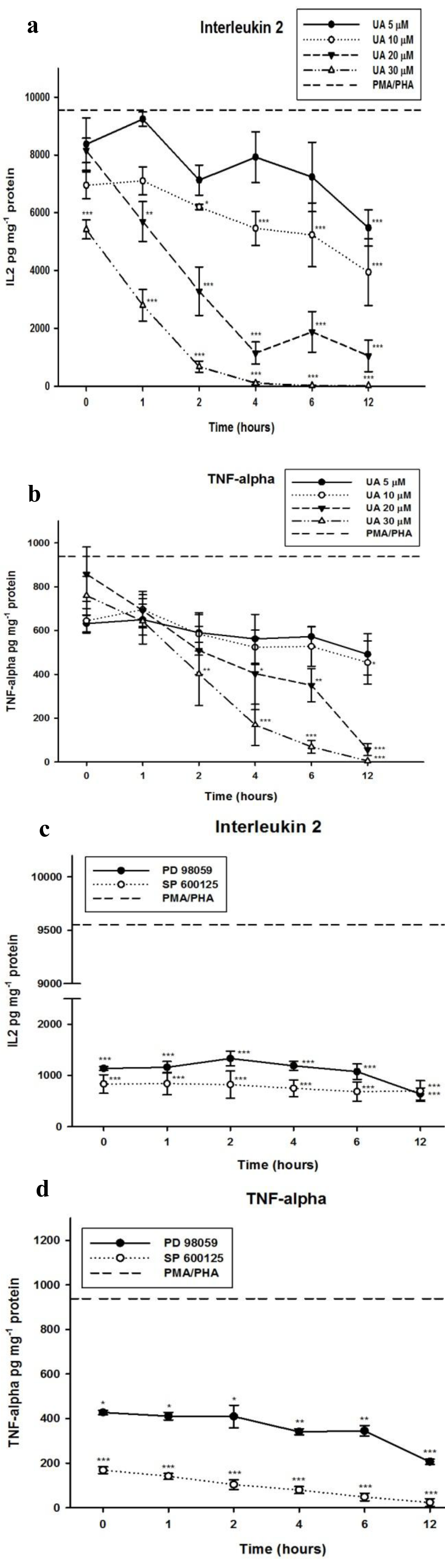
Figure 3: Effect of UA and MAPK inhibitors on inhibition of PMA/PHA-induced IL-2 and TNF-α production. Cells were treated with DMSO (control), UA, PD98059 or SP600125 for 0 to 12 hours and then stimulated with PMA/PHA for a further 12 hours. The IL-2 (a), (c) and TNF-α (b), (d) were determined using ELISA. Results are expressed as mean SEM from three independent experiments. Statistical analyses were carried out with one-way ANOVA followed by Tukey's multiple comparison test compared with positive controls (PMA/PHA) (* P < 0.05, ** P < 0.01, *** P < 0.001).
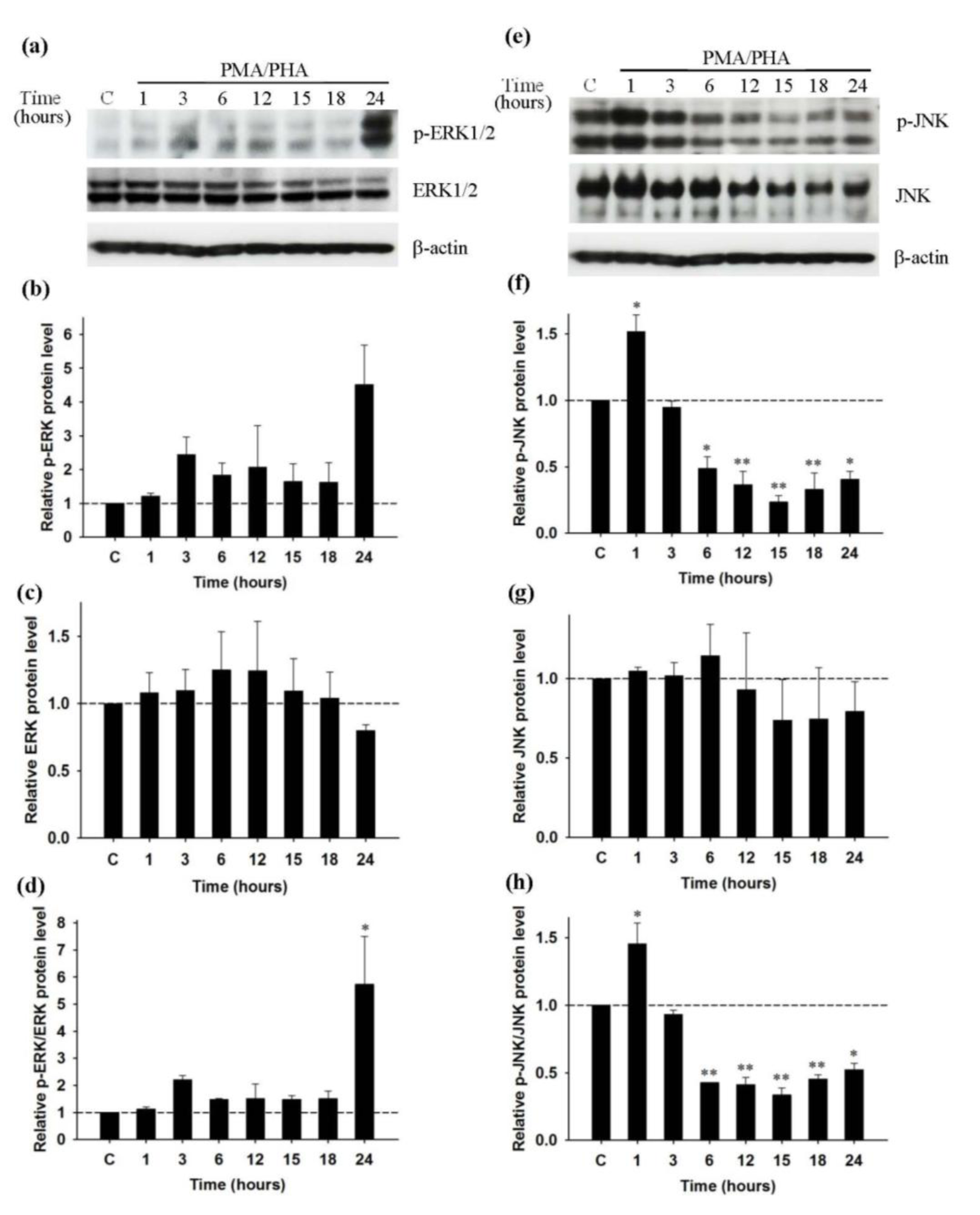
Figure 4: Effect of PMA/PHA on the activity of ERK1/2 and JNK in Jurkat leukemic T-cells; Western blot analysis (representative figure of three independent experiments). Cells were incubated with PMA/PHA for 1 to 24 hours, or only DMSO (C) for 24 hours. Protein expression of (a) p-ERK1/2 (42, 44 kDa), ERK1/2 (42, 44 kDa) and β-actin (45kDa) was detected by Western blot and normalized with β-actin. (b), (c), (d) Relative p-ERK, ERK and p-ERK/ERK protein level was calculated. Protein expression of (e) p-JNK (46, 54 kDa), JNK (46, 54 kDa) and β-actin (45kDa) was detected by Western blot and normalized with β-actin. (f), (g), (h) Relative p-JNK, JNK and p-JNK/JNK protein level was calculated. Statistical analysis was carried out with one-way ANOVA followed by Tukey's multiple comparison test compared with positive controls (PMA/PHA) (* P < 0.05, ** P < 0.01)
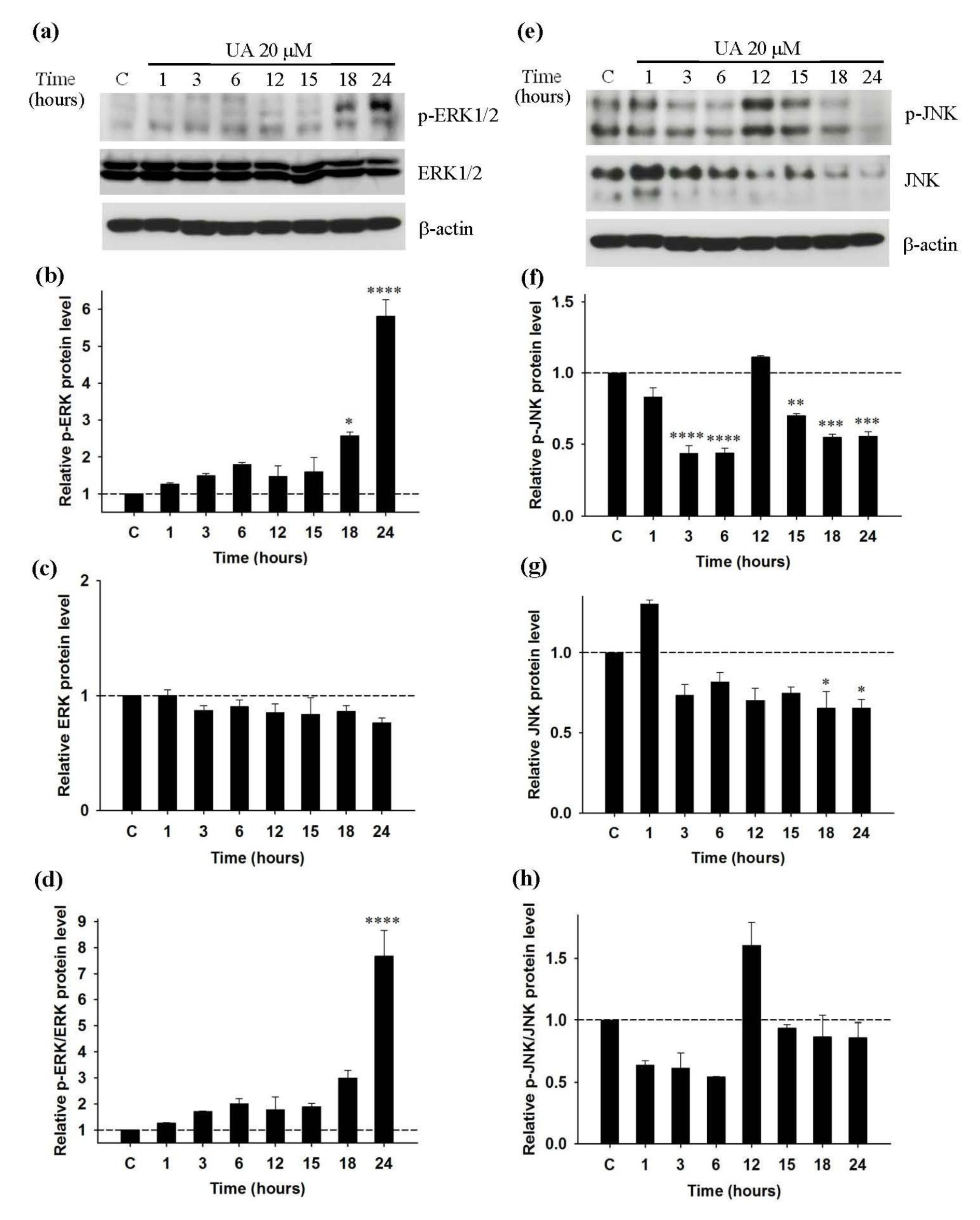
Figure 5: Effect of UA on the activity of ERK1/2 and JNK in Jurkat leukemic T-cell; Western blot analysis (representative figure of three independent experiments). Cells were incubated with 20 µM UA for 1 to 24 hours, or only DMSO (C) for 24 hours. Protein expression of (a) p-ERK1/2 (42, 44 kDa), ERK1/2 (42, 44 kDa) and β-actin (45kDa) was detected by Western blot and normalized with β-actin. (b), (c), (d) Relative p-ERK, ERK and p-ERK/ERK protein levels were calculated. Protein expression of (e) p-JNK (46, 54 kDa), JNK (46, 54 kDa) and β-actin (45kDa) were detected by Western blot and normalized with β-actin. (f), (g), (h) Relative p-JNK, JNK and p-JNK/JNK protein levels were calculated. Statistical analysis was carried out with one-way ANOVA followed by Tukey's multiple comparison test compared with positive controls (PMA/PHA) (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001).
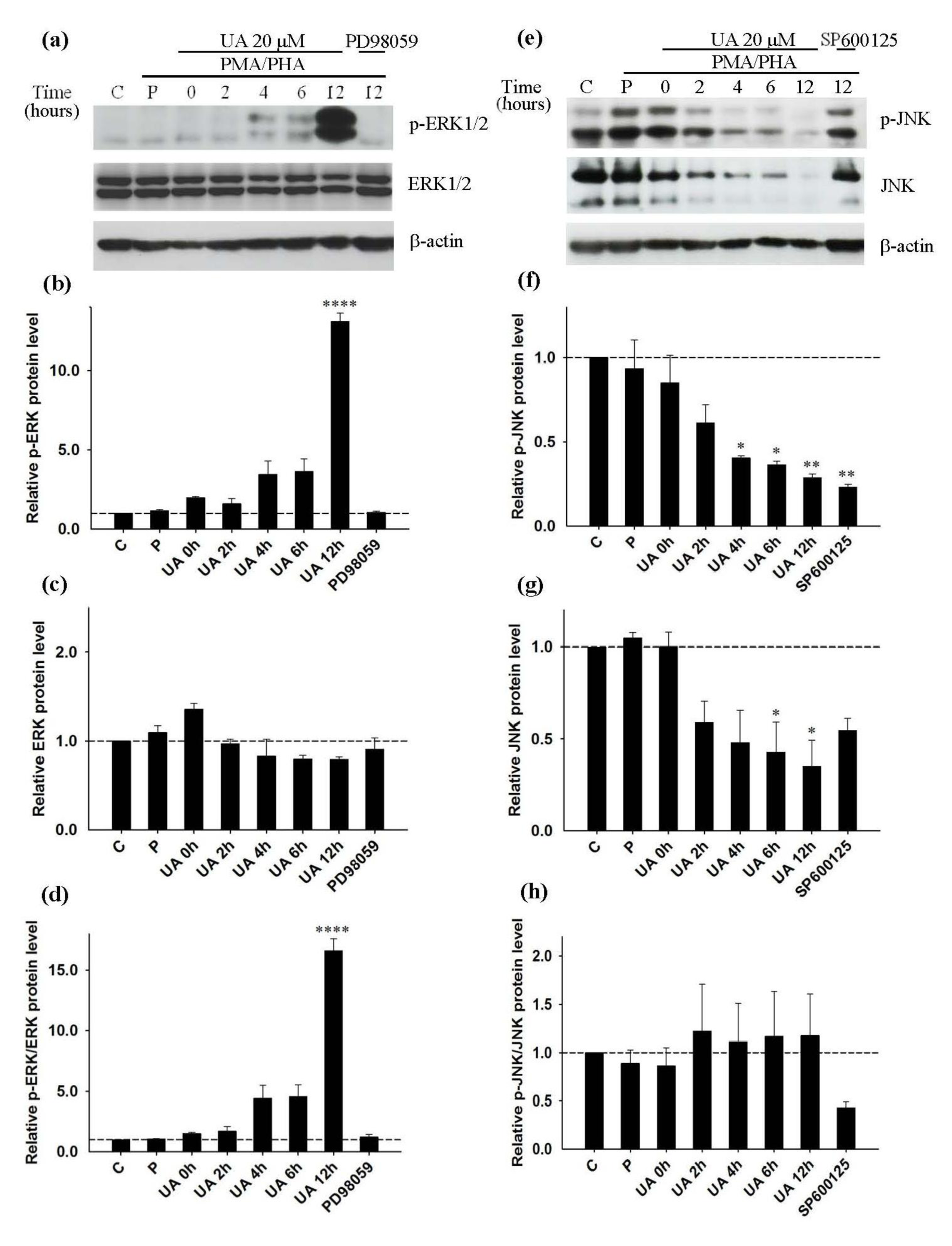
Figure 6: Inhibition of PMA/PHA induced-IL-2 and TNF-α production by ursolic acid via the JNK pathway; Western blot analysis (representative figure of three independent experiments). Cell were pre-incubated with UA for 0 to 12 hours, PD98059 or SP600125 for 12 hours prior to addition of PMA/PHA for a further 12 hours, or only DMSO (C) for 24 hours. For positive control (P) cells were left without treatment for 12 hours prior to addition of PMA/PHA for a further 12 hours. Protein expression of (a) p-ERK1/2 (42, 44 kDa), ERK1/2 (42, 44 kDa) and β-actin (45kDa) was detected by Western blot and normalized with β-actin. (b), (c), (d) Relative p-ERK, ERK and p-ERK/ERK protein level was calculated. Protein expression of (e) p-JNK (46, 54 kDa), JNK (46, 54 kDa) and β-actin (45kDa) was detected by Western blot and normalized with β-actin. (f), (g), (h) Relative p-JNK, JNK and p-JNK/JNK protein level was calculated. Statistical analysis was carried out with one-way ANOVA followed by Tukey's multiple comparison test compared with positive controls (PMA/PHA) (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001).
[*] Corresponding Author:
Dr Sirikalaya Brimson, Center for Research and Development in Molecular Hematology Sciences, Department of Clinical Microscopy, Faculty of Allied Health Sciences, Chulalongkorn University, Bangkok 10330, Thailand; Tel.: +66-2218-1086 ext. 332; Fax: +66-2-218-3771, eMail: Sirikalaya.J@chula.ac.th, Peach10230@yahoo.com