Research article
Coriandrum sativum L. seed extract mitigates lipotoxicity in RAW 264.7 cells and prevents atherogenic changes in rats
Dipak Patel1, Swati Desai1, Tejal Gajaria1, Ranjitsinh Devkar1[*], A.V. Ramachandran1
1Division of Phytotherapeutics and Metabolic Endocrinology, Department of Zoology, Faculty of Science, The M. S. University of Baroda, Gujarat, IndiaEXCLI J 2013;12:Doc313
Abstract
This study was designed to assess the efficacy of Coriandrum sativum L. (CS) in preventing in vitro low density lipoprotein (LDL) oxidation mediated macrophage modification. Further, an in vivo study was also conducted to confirm upon the efficacy of CS seed extract in alleviating pathophysiological alterations of high cholesterol diet induced atherosclerosis in rats. Copper mediated cell free oxidation of LDL accounted for elevated indices of malondialdehyde (MDA), lipid hydroperoxide (LHP)and protein carbonyl (PC) and a progressive increment in conjugate diene (CD) levels whereas, reverse set of changes were recorded in presence of CS extract. Cell mediated LDL oxidation (using RAW 264.7 cells) accounted for lowered MDA production and oxidized LDL (Ox-LDL) mediated cell death in presence of CS extract and the same was attributed to its potent antioxidant and free radical scavenging potentials. High cholesterol fed atherogenic rats showed elevated lipid indices, evidences of LDL oxidation, plaque formation in thoracic aorta. The same was further validated with immunostaining of cell adhesion molecules and hematoxylin and eosin (HXE) staining. However, co-supplementation of CS to atherogenic rats recorded significant lowering of the above mentioned parameters further strengthening the claim that CS extract is instrumental in preventing onset and progression of atherosclerosis.
Keywords: Coriandrum sativum L., Hydro-methanolic seed extract, LDL oxidation, atherosclerosis
Introduction
Hyperlipidemia is a major cause of cardiovascular disorders such as atherosclerosis and coronary heart disease (Chobanian, 1991[16]). High levels of circulating cholesterol have been identified as a potential risk factor for atherosclerosis and coronary heart disease (Kannel et al., 1979[38]). World Health Organization has predicted that, almost 23.6 million people will die due to cardiovascular diseases (CVD) by 2030 (Halliwell, 1995[28]). Hence, hypercholesterolemia characterized by elevated plasma low-density lipoprotein cholesterol (LDL) levels is an important risk factor for the development and progression of atherosclerosis (Kannel et al., 1975[39]; Keys, 1970[41]) wherein, oxidative modification of LDL plays a pivotal role (Heinecke, 1997[31]). Formation of oxidized LDL (Ox-LDL) is triggered by transition metals, plasma enzymes, vascular endothelium or smooth muscle cells (Sparrow and Olszewski, 1993[67]). It has been reported that the oxidatively modified LDL is cytotoxic, chemotactic and chemostatic (Napoli et al., 1997[50]; Steinberg, 1997[68]). LDL particles (< 70 nm) are more prone to enter the tunica intima of thoracic aorta and undergo cell mediated oxidation (Simionescu and Simionescu, 1993[66]). This process is coupled with expression of cell adhesion molecules on vascular endothelium to trigger recruitment of monocyte that soon differentiate into macrophages (Glass and Witztum, 2001[25]). At this state, the macrophages exhibit scavenger receptor mediated Ox-LDL uptake and subsequently get transformed into lipid laden foam cells. These cells soon undergo apoptosis to form apoptotic bodies that initiate formation of fatty streak. As a result, more macrophages are recruited and undergo similar fate and contribute towards formation of an atherosclerotic lesion (Björkerud and Björkerud, 1996[7]; Harada-Shiba et al., 1998[29]). Since, the sequence of events is irreversible and hence, reduction in the level of triglyceride, cholesterol and LDL by naturally occurring compounds is often considered as an effective alternative in alleviating development of an atherosclerostic lesion (Novo et al., 2011[53]).
Coriandrum sativum L. (Apiaceae) (CS) is an annual herb, the fresh leaves and dried seeds of which a common component of Middle Eastern, Mediterranean, Indian, Latin American, African and Southeast Asian cuisines. Decoction and tincture of powdered seeds of CS alone or in combination with other herbal agents are recommended for dyspeptic complaints, loss of appetite, convulsion, insomnia and anxiety (Grieve, 1971[27]). It is also used as medication against diabetes, indigestion, flatulence, renal disorders and as a diuretic agent in India and Morocco (Grieve, 1971[27]; Emamghoreishi et al., 2005[20]). It is also used in the treatment of urethritis, cystitis, urinary tract infection, urticaria, rashes, burns, sore throat, vomiting, indigestion, nosebleed, cough, allergies, hay fever, dizziness and amoebic dysentery (Grieve, 1971[27]; PDR, 2004[55]).
Phytochemical constituents of CS seeds have been studied extensively and their analysis has revealed the presence of polyphenols (rutin, caffeic acid derivatives, ferulic acid, galic acid, and chlorogenic acid), flavonoids (quercetin and isoquercetin) and β-carotenoids (Melo et al., 2003[48]). The essential oil obtained from CS seeds contains α and β-pinene, camphor, citronellol, coriandrol, p-cymene, geraniol, geranyl acetate, limonene, linalool, myrcene, α and β phellandrene and α and β-terpinene along with many fatty acids, a large number of water soluble compounds have been identified including monoterpenoid glycosides, monoterpenoid glucose sulfate and other glycosides (Sergeeva, 1975[63]; Ishikawa et al., 2003[35]). The pharmacological activities of various extracts and the essential oil of CS seeds have been also studied. The essential oil has been found to possess antimicrobial (Baratta et al., 1998[6]) and antifungal properties (Garg and Siddiqui, 1992[23]). Its efficacy as a hypoglycemic (Gray and Flatt, 1999[26]), hypolipidemic (Chithra and Leelamma, 1997[15], 1999[14]; Lal et al., 2004[43]), hypocholesterolemic (Dhanapakiam et al., 2008[19]), antihypertensive (Medhin et al., 1986[47]), antioxidant (Melo et al., 2003[48]; Ramadan et al., 2003[58]; Bajpai et al., 2005[5]), antimutagenic (Cortes-Eslava et al., 2004[18]), anxiolytic (Emamghoreishi et al., 2005[20]), antimicrobial (Kubo et al., 2004[42]; Cantore et al., 2004[11]), larvicidal (Consoli et al., 1988[17]) and post-coital antifertility (Al-Said et al., 1987[2]) agent have also been reported.
Hashim et al. (2005[30]) reported that polyphenol rich fractions of CS possess strong anti-oxidant potential and an ability to alleviate hydrogen peroxide (H2O2) mediated oxidative damage in human lymphocytes. Also, CS administration resulted in preventing hyperlipidemia in high fat diet fed rats (Chithra and Leelamma, 1997[15]). Owing to these credentials of CS seed extract, the present study was designed to assess its efficacy in preventing in vitro LDL oxidation mediated macrophage modification. Also, the in vivo study is aimed at further confirmation of the efficacy of CS seed extract in alleviating pathophysiological alterations in high cholesterol diet fed atherogenic rats.
Material and Methods
Plant material and preparation of extract
CS plants were collected in the seedling months (February and March) and Dr. P.S. Nagar, Department of Botany, The M.S. University of Baroda identified the plant and deposited in the herbarium of the Department of Botany. One hundred grams of powdered dry seeds were soaked in methanol:water (80:20 v/v) at room temperature and allowed to stand for seven days. The resultant extract was filtered through a muslin cloth and then concentrated in a rotary evaporator under reduced pressure to obtain a thick semisolid brown paste (Hashim et al., 2005[30]). The final yield was 8.3 g (w/w).
In vitro study
Isolation of LDL
Venous blood was collected by a pathologist from fasting healthy volunteers with normal levels of cholesterol as per the standard guidelines. These samples were kept at room temperature for 45 min and serum was obtained by centrifuging at 3000 rpm for 10 min at 4 °C. LDL was isolated using heparin-citrate buffer (64 mM trisodium citrate at pH 5.05 containing 50,000 IU/l heparin) precipitation method (Ahotupa et al., 1998[1]). A mixture of 0.1 ml of serum and 1 ml of the heparin-citrate buffer was vortexed and allowed to stand for 10 min at room temperature and later centrifuged (3,000 rpm for 10 min at 20 °C) to remove insoluble lipoproteins. The resultant pellet was suspended in 0.1 ml of phosphate-buffered saline (PBS; 0.1 M, pH 7.4, containing 0.9 % sodium chloride (NaCl)). Protein concentration of the LDL was estimated by the method of Lowry et al. (1951[46]) using bovine serum albumin as standard.
LDL oxidation kinetics
0.1 ml of LDL (100 μg protein) was diluted to 0.9 ml with PBS and was incubated in presence or absence of 0.1 ml of CS extract (10-100 μg/ml) at 37 °C for 30 min. Later, freshly prepared 0.167 mM Copper sulfate (CuSO4) solution was added to initiate LDL oxidation. The LDL oxidation kinetics was determined by continuously monitoring (every 10 min) the absorbance for 180 min (at 37 °C) at 234 nm in a UV/VIS Perkin Elmer spectrophotometer. Lag time (min) was determined from the intercepts of lines drawn through the linear portions of the lag phase and propagation phase. The rate of oxidation was determined from the slope of the propagation phase. The concentration of conjugate diene (CD) in the samples was calculated by using a molar extinction coefficient of 2.95 x 104 M-1cm-1. Maximum concentration of CD formed was calculated from the difference in the concentration of CD at zero time and at diene peak (absorption maxima) (Esterbauer et al., 1989[21]).
LDL oxidation products
Three sets of tubes were prepared to assess the rate of LDL oxidation in presence or absence of CS extract. Copper-mediated LDL oxidation was carried out in presence or absence of CS extract (10-100 μg/ml) for 24 h as mentioned above. Later, 0.01 ml of 10 mM ethylenediaminetetraacetic acid (EDTA) was added in each tube to stop oxidation reaction and each sample was processed for measurement of malondialdehyde (MDA), lipid hydroperoxide (LHP) and protein carbonyl (PC) as follows:
Malondialdehyde: 0.1 ml of aliquot was mixed with 1 ml thiobarbituric acid (TBA) reagent (0.37 % TBA, 15 % trichloroacetic acid (TCA) in 0.25 N hydrochloric acid (HCl) and placed in water bath at 100 °C for 30 min, cooled to room temperature and centrifuged at 3000 rpm for 10 min. The absorbance of the supernatant was measured at 532 nm with UV-VIS Perkin Elmer spectrophotometer and MDA was calculated using a molar extinction coefficient of 1.56 x 105 M-1cm-1 (Buege and Aust, 1978[9]).
Lipid hydroperoxide: 0.1 ml of aliquot was mixed with 0.9 ml of Fox reagent (0.25 mM ammonium sulphate, 0.1 mM xylenol orange, 25 mM sulfuric acid (H2SO4), and 4 mM Butylated hydroxytoluene (BHT) in 90 % (v/v) HPLC-grade methanol) and incubated at 37 °C for 30 min. The absorbance was read at 560 nm and LHP content was determined using the molar extinction coefficient of 4.3 x 104 M-1 cm-1 (Nourooz-Zadeh et al., 1996[52]).
Protein carbonyls: 0.1 ml of aliquot was mixed with 0.2 ml of 2,4-Dinitrophenylhydrazine (DNPH) (in 2 M HCl). After incubation at room temperature for 60 min, 0.6 ml of denaturing buffer (150 mM sodium phosphate buffer containing 3 % sodium dodecyl sulfate (SDS) was added and mixed thoroughly. Later, ethanol and heptane (1.8 ml of each) to precipitate protein and the contents were centrifuged. The protein was washed three times with 1.5 ml of ethyl acetate/ethanol (1:1, v/v), dissolved in 1 ml of denaturing buffer, and read at 360 nm in a spectrophotometer. The carbonyl content was calculated using molar extinction absorption coefficient of 22.0 x 103 M-1cm-1 (Reznick and Packer, 1994[60]).
Apolipoprotein B100 (ApoB) fragmentation
Copper-mediated LDL oxidation was carried out in presence or absence of CS extract (10-100 μg/ml) for 24 h as mentioned above. Later, 0.01 ml of 10 mM EDTA was added in each tube to stop oxidation. Samples were centrifuged and LDL obtained was denatured with 3 % SDS, 10 % glycerol, and 5 % bromophenol at 95 °C for 5 min and cooled to room temperature. Later, LDL samples were loaded on 8 % SDS-polyacrylamide gels, and electrophoresis was performed at 100 V for 60 min. The gels were stained with 2 % coomassie brilliant blue solution for 6 h at 4 °C and de-stained (20 % glacial acetic acid and 10 % methanol in water) for 30 min. Later, gels were cleared in 15 and 10 % acetic acid for 10 min each, fixed in 10 % glycerol and photographed using canon power shot S70 digital camera (Lee et al., 2002[44]).
Relative Electrophoretic Mobility (REM)
LDL was subjected to copper ion (Cu2+) mediated oxidation in presence or absence of CS extract (10-100 μg/ml) for 24 h as mentioned above. Later, 0.01 ml of 10 mM EDTA was added in each tube to stop oxidation and the contents were centrifuged to obtain a pellet. The electrophoretic mobility of native or oxidized LDL (with or without CS extract) was detected using agarose gel electrophoresis (Reid and Mitchinson, 1993[59]). Samples were loaded on 0.6 % agarose gel and electrophoresed (100 V) in 50 mM barbituric acid (pH 8.6) for 40 min. After electrophoresis, the gels were fixed in a solution containing 60 % methanol, 30 % water and 10 % glacial acetic acid for 30 min, dried at 50 °C for 40 min and stained with 0.6 % Sudan black B for 60 min. Gels were photographed and the results were expressed in terms of distance (meter) travelled by LDL from origin.
Preparation of oxidized LDL (Ox-LDL) and culture of RAW 247.6 cells
0.1 ml of LDL (100 μg protein) was diluted to 0.9 ml with PBS and incubated for 24 h at 37 °C. LDL was oxidized with 0.01 ml freshly prepared CuSO4 (0.167 mM). Analysis of MDA and CD were carried out in the LDL samples. Samples with MDA 50 ± 5 nmol/mg LDL protein and CD 80 ± 8 nmol/mg LDL protein were used for further studies. RAW 247.6 cells (Macrophages) were purchased from National Centre of Cell Sciences, Pune, India. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10 % Fetal bovine serum (FBS) and 1 % antibiotic-antimycotic solution in a humidified incubator at 37 °C with 5 % CO2.
Cell mediated LDL oxidation
RAW 247.6 cells (1 x 105/ml) were incubated in 1 ml of Ham's F-12 medium (without phenol red) containing LDL (100 μg/ml) at 37 °C for 24 h. Cell-free control well was used for all conditions. At the end of incubation, oxidation was arrested by chilling the medium and adding 0.2 mM EDTA and 0.04 mM BHT. Later, 0.1 ml of each supernatant was used for the assay of MDA described earlier (Thounaojam et al., 2011[71]).
Ox-LDL induced foam cell formation
RAW 247.6 cells treated with CS extract (100 μg/ml) were incubated in presence of 100 μg/ml of Ox-LDL for 24 h. Later, medium was decanted and cells fixed in 4 % paraformaldehyde for 15 min. The cells were then washed twice with PBS, and stained in 1 % Oil red O solution for 30 min. At the end of staining, excess Oil red O was removed and 1 ml of glycerin added. Photographs were taken on Leica DMIL inverted microscope using canon power shot S 70 digital camera (Jadeja et al., 2011[36]).
Intracellular oxidative stress
Macrophage cells (1 x 105/ml) treated with CS (100 μg/ ml) were incubated in presence of 100 μg/ ml of Ox-LDL for 24 h. Later, the cells were further incubated for 60 min at 37 °C and incubated with 0.0075 mM 2',7'-dichlorfluorescein-diacetate (DCF-DA) for 30 min in dark (Silva et al., 2010[65]). Photographs were taken using canon power shot S70 digital camera in Leica DMRB florescence microscope.
Mitochondrial Membrane Potential (MMP) assay
MMP in control and CS treated cells was measured using a fluorescent cationic dye Rhodamine123 (RHO123) (Pereira and Oliveira, 2000[56]). RAW 247.6 cells (1 x 105/ml) pretreated with CS extract (10-100 μg/ml) for 30 min were incubated in presence of 100 μg/ml of Ox-LDL for 24 h. The cells were then incubated with 0.001 mM RHO123 for 10 min at 37 °C and the fluorescence was determined (485 and 530 nm excitation and emission respectively) using spectroflurometer (Jasco FP-6350).
Cytotoxicity assay
RAW 247.6 cells (1 x 104) pretreated with CS extract (10-100 μg/ml) for 30 min were incubated in presence of 100 μg/ml of Ox-LDL for 24 h. Further incubation of the cells was carried out in a culture medium containing 0.5 mg/ml 3-(4,5-Dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide (MTT) for 160 min. Later, 0.15 ml of dimethyl sulphoxide was added to all the wells and was incubated for 30 min at room temperature with constant shaking. Absorbance was read at 540 nm using ELX800 Universal Microplate Reader (Bio-Tek instruments, Inc, Winooski, VT) and % cell viability was calculated.
Nuclear condensation study
Control or pre-treated (CS extract 100 μg/ml for 30 min) RAW 247.6 cells (1 x 105/ml) were incubated with Ox-LDL (100 μg/ml) of for 24 h. A single-cell suspension of treated cells was washed in PBS, fixed in 70 % ethanol. Cells were washed again with PBS and incubated with DAPI stain (0.6 μg/ml in PBS) for 5 min. Chromatin fluorescence was observed under a Leica DMRB 2000 fluorescence microscope. Apoptotic cells were morphologically defined by cytoplasmic and nuclear shrinkage and chromatin condensation (Hsieh et al., 2007[32]).
Cell cycle analysis
RAW 247.6 cells (1 x 106/ml) were cultured in presence of CS extract (100 μg/ml) and 100 μg/ml of Ox-LDL for 24 h. At the end of incubation, cells were collected, washed twice with PBS, fixed overnight in cold 70 % ethanol at 4 °C and resuspended in PBS. Cells were incubated with RNase A for 45 min and stained with propidium iodide (1 mg/ml) in the dark at 37 °C for 30 min (Pozarowski and Darzynkiewicz, 2004[57]). The suspension was analyzed with on Flow Cytometer (BD FACSAria III, USA). The apoptosis was determined based on the “sub-G1” peak.
In vivo studies
Experimental animals
Age matched (9-10 weeks old) male Sprague Dawley rats weighing 300 ± 20 gm (Zydus research centre, Ahmedabad) were maintained in clean polypropylene cages and fed with laboratory chow (M/S Pranav agro, Ltd. Baroda, India) and water ad libitum. The experimental protocol was executed according to the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), India and approved by the animal ethical committee of the Department of Zoology, The M.S. University of Baroda, Vadodara (Approval No.827/ac/04/CPCSEA).
Induction of atherosclerosis
In the present study, a total of 24 rats were divided into three groups of eight animals each. Group I: CON; rats were given single dose of 0.9 % saline (0.1 ml) intraperitoneally and maintained on standard laboratory chow and simultaneously administered with 0.5 % carboxymethyl cellulose (CMC) (0.1 ml) orally for 8 weeks. Group II: ATH; rats were given single dose of Vitamin D3 (600,000 IU unit/kg) intraperitoneally and later, fed with an atherosclerotic diet (3 % cholesterol, 0.5 % cholic acid, 0.2 % 6-propyl 2-thiouracil, 5 % sucrose, 10 % lard, and 81.3 % powdered laboratory chow) and simultaneously administered with 0.5 % CMC (0.1 ml) orally for 8 weeks. Group III: ATH+CS; rats were given single dose of Vitamin D3 (600,000 IU unit/kg) intraperitoneally and later fed with an atherosclerotic diet and simultaneously administered with 200 mg/kg of CS extract by oral feeding for 8 weeks (Cai et al., 2005[10]; Huang et al., 2004[33]). At the end of the experimental period, blood was collected from overnight fasted rats (12 h) via retro-orbital sinus puncture under mild ether anaesthesia in 2 ml centrifuge tubes and serum was separated by cold centrifugation (4 °C) at 1500 rpm for 10 min. Later, animals were sacrificed by cervical dislocation under mild ether anaesthesia and thoracic aorta of control and experimental animals were collected. Two small pieces of thoracic aorta from aortic arch were collected and processed for paraffin wax histology. The remaining piece of thoracic aorta was stored at -80 °C (Cryo Scientific Ltd., India) for further use.
Serum lipids
Serum triglyceride (TG), total cholesterol (TC) and high density lipoprotein (HDL) contents were estimated with commercially available enzymatic kits (Reckon Diagnostics Ltd., Baroda, India) using a semi auto-analyser (Micro lab 300 L, Merck) and levels of LDL and very low density lipoprotein (VLDL) were calculated (Friedewald et al., 1972[22]).
Isolation of LDL from rats and MDA assay
LDL was isolated from serum samples of control and experimental rats by heparin-citrate buffer precipitation method as described earlier (Ahotupa et al., 1998[1]). The protein concentration of LDL was estimated by the method of Lowry et al. (1951[46]) using BSA as standard. Oxidation state was evaluated by assaying MDA levels in the LDL samples of control and experimental groups as mentioned above and the absorbance was measured at 532 nm with UV/VIS Perkin Elmer spectrophotometer and, MDA was calculated using a molar absorption coefficient of 1.56 × 105 M-1cm-1 (Buege and Aust, 1978[9]).
Microscopic evaluation of thoracic aorta
Thoracic aorta of control and experimental rats were fixed in 4 % buffered paraformaldehyde, dehydrated in graded alcohol series and embedded in paraffin wax using automated tissue processor. Five to seven μm sections were cut (on a Leica RM 2155 Microtome), stained with hematoxylin and eosin (HXE). Another set of sections of aorta from control and experimental rats was incubated in von Kossa stain solution (1 % silver nitrate) under ultraviolet light for 20 min. The sections were then rinsed repeatedly in distilled water, placed in 5 % sodium thiosulphate for 5 min and rinsed in distilled water again (to remove unreacted silver). The sections were then counterstained with 1 % eosin for 5 min (Hsieh et al., 2007[32]). All sections were examined under a Leica DMRB microscope (100 X) and photographed with a canon power shot S70 digital camera at 100 X magnification.
Immunohistochemistry of thoracic aorta
Paraffin embedded sections of thoracic aorta of control and experimental rats were deparaffinised in xylene and hydrated using graded series of alcohol and water. Sections were then washed in phosphate buffer saline (PBS) and antigen retrieval step was carried out by immersing slides in sodium citrate buffer at 80 °C for 10 min. Later, endogenous peroxidases were removed by incubation of sections in 3 % H2O2 for 20 min in dark. Non-specific binding sites were blocked by incubation of slides with 1 % FBS for 30 min. Localization of vascular cell adhesion molecule-1 (VCAM-1), and P-selectin was carried out by incubating sections with rabbit anti-rat IgG at a dilution of 1:100 (SantaCruz Biotechnology, Inc.) and goat anti-rat P-selectin IgG at a dilution of 1:100 (Santa Cruz Biotechnology,Inc.), respectively for overnight at 4 °C in a humidified chamber. At the end of incubation, slides were washed with PBS and then, the sections were incubated with respective horseradish peroxidise (HRP) conjugated secondary antibodies for 4 h at room temperature. Goat anti-rabbit IgG-HRP 1:100 (Bangalore Genei Pvt Ltd.) for VCAM-1, and rabbit anti-goat IgG-HRP 1:100 (Bangalore Genei Pvt Ltd.) for P-selectin were used. At the end of incubation, sections were thoroughly washed with PBS and final detection step was carried out using diaminobenzidine (DAB) detection system (Bangalore Genei Pvt Ltd.) and counterstained with haematoxylin. Sections were examined under Leica DMRB microscope and photographed using a canon Power shot S70 digital camera (Thounaojam et al., 2012[72]).
Statistical analysis
Statistical evaluation of the data was done by one-way ANOVA followed by Bonferroni's multiple comparison tests. The results were expressed as mean ± S.E.M using Graph Pad Prism version 3.0 for Windows, Graph Pad Software, San Diego, CA/USA.
Results
In vitro study
LDL oxidation kinetics
LDL subjected to treatment with 10 μM CuSO4 showed significantly (p<0.001) increased CD formation and reduced lag time. In contrast, CS (25, 50, 75, and 100 μg/ml) was able to reduce CD formation and delay lag time in a dose-dependent manner (Figure 1(Fig. 1)).
LDL oxidation products (MDA, LHP, and PC)
There was significant (p<0.001) elevation in formation of MDA, LHP and PC after treatment of LDL with 10 μM CuSO4 as compared to the control group. However, presence of CS (25, 50, 75, and 100 μg/ml) accounted for a dose dependent reduction in formation of LDL oxidation products (Figure 2(Fig. 2)).
Relative Electrophoretic Mobility (REM) and ApoB fragmentation
The degree of CuSO4 mediated LDL oxidation was assessed by the extent of altered REM and ApoB fragmentation.
There was significant increment in electrophoretic mobility in Ox-LDL whereas; the same showed a dose dependent reduction following CS treatment. Also, the Ox-LDL showed fragmentation of ApoB and the same was evidenced by complete absence of the band in electrophoretic pattern observed herein.
However, CS (10, 25, 50, 75, and 100 μM) treatment accounted for reappearance of the ApoB band in a dose dependent manner (Figures 3a and 3b(Fig. 3)).
Cell mediated LDL oxidation
Oxidation of LDL mediated by RAW 247.6 cells recorded significantly (p<0.001) increased MDA levels as compared to the control group whereas, presence of CS (10, 25, 50, 75, and 100 μM) accounted for a decrement in MDA levels with highest doses being most significant (Figure 4(Fig. 4)).
Foam cells
Incubation of RAW 247.6 cells with Ox-LDL (100 μg/ml) for 24 h resulted in significant uptake of Ox-LDL, leading to higher intracellular cholesterol accumulation compared to Ox-LDL deprived cells. Addition of CS extract to Ox-LDL treated macrophages significantly reduced intracellular cholesterol accumulation (Figure 5(Fig. 5)).
Intracellular oxidative stress
As shown in Figure 6(Fig. 6) Ox-LDL (100 μg/mL) treated RAW 247.6 cells showed a visible increment in the intensity of green color. The same is indicative of high content of ROS generated and elevated levels of intracellular oxidative stress. However, presence of CS (100 μg/mL) was instrumental in recording observable decrement in the intensity of green color within the cytoplasm of cells.
Mitochondrial membrane potential and cytotoxicity assay
RAW 247.6 cells treated with Ox-LDL (100 μg/mL) showed a significant decrement (p<0.001) in cell viability and mitochondrial membrane potential whereas; the same showed a dose dependent improvement following CS treatment (Figures 7(Fig. 7) and 8(Fig. 8)).
Nuclear condensation and cell cycle analysis
Cells incubated with Ox-LDL were stained with DAPI and PI showed typical characteristics of chromatin condensation and accumulation of cells in sub-G0 phase. However, CS treatment recorded visibly less number of cells with condensed nuclei. Also, the cell cycle analysis of Ox-LDL+CS treated cells accounted for significantly less number of cells in G0 phase suggesting improved cell survival (Figures 9(Fig. 9) and 10(Fig. 10)).
In vivo study
Serum lipid profile and MDA assay
There was significant (p<0.001) elevation in serum TC, TG, VLDL and LDL levels and, a decrement in HDL level recorded in ATH rats as compared to the control rats. However, ATH+ CS rats showed significant (p<0.001) decrement in TC, TG, VLDL and LDL levels and increment in HDL level as compared to ATH group. There was significant (p<0.001) increment in MDA level the LDL isolated from ATH rats as compared to the control rats. However, ATH+CS treated rats recorded significant (p<0.001) decrement in MDA level (Figures 11(Fig. 11) and 12(Fig. 12)).
Microscopic evaluation of thoracic aorta
The photomicrographs of thoracic aorta of control rats stained with hematoxylin and eosin showed normal histo-architecture with intact intima and linear pattern of smooth muscles in all observed tissue sections. However, there was a significant distortion of intima, smooth muscle derangement, degenerative changes in media and atheromatous plaque formation observed in ATH rats. The ATH+CS rats showed relatively less distortion of intima and smooth muscles with small patches of atheromatous plaques as compared to ATH rats. The von Kossa stained sections of aorta showed a high level of calcification in ATH rats as compared to control rats whereas, the same showed minimal calcification in aorta of ATH+CS treated rats (Figures 13a and 13b(Fig. 13)).
Immunohistochemical (IHC) localization of adhesion molecules in aorta
Cell adhesion molecules (CAMs; VCAM-1 and p-selectin) were immunolocalized in section of thoracic aorta of control and treated rats. The endothelial lining of aorta of control rats showed no evidence of immunostaining of both CAMs but the same were visibly intense in ATH rats. However, ATH+CS rats recorded lower grade of expression and the same was evident in their stained sections (Figures 14a and 14b(Fig. 14)).
Discussion
Atherosclerosis is a multifactorial disease wherein hypercholesterolemia and oxidative modification of LDL are the key initiation factors in its development and progression (Zha et al., 2009[76]). Oxidative modification in n-LDL in vivo results from an imbalance between the pro-oxidant challenges and antioxidant defences (Moriel et al., 2002[49]). Various in vitro methods have been developed to evaluate the contributions exerted by intrinsic factors in triggering oxidative modifications of LDL particles (Lobato et al., 2009[45]). In the present study, LDL was isolated from human serum by precipitation method and subsequently challenged with copper ion so as to obtain Ox-LDL. The kinetics of LDL oxidation was subsequently evaluated by assessing its oxidation products such as CS whereas; the non-kinetic indices such as MDA, LHP and PC have also been assessed (Thounaoujam et al., 2011[71]). These parameters have been extensively used by various research groups to establish the efficacy of a drug or herbal extract in preventing LDL oxidation (Thounaoujam et al., 2011[71], Yu et al., 2005[75], Visavadiya et al., 2009[73]). In the present study Cu+2 treatment accounted for elevated indices of MDA, LHP and PC a progressive increment in CD levels. These results confirm Cu+2 mediated oxidation of LDL. These results are in accordance with studies performed by other research group as well as previous studies from our lab (Thounaoujam et al., 2012[72]; Jadeja et al., 2012[37]). Presence of CS extract accounted for significant decrement in LDL oxidation products. The same was evidence in form of increased lag time (100 min) as compared to Cu+2 mediated LDL oxidation. It is assumed that the high polyphenol content present in CS extract scavenges the free radicals thus accounting for a prolonged lag time and significant decrement in formation of the LDL oxidation products.
Fragmentation of fatty acids during atherogenic progression is associated with generation of highly reactive intermediates such as aldehydes and ketones that are instrumental in causing imperative modifications of LDL. This is known to oxidatively delete peptide bonds and cause derivatization of the lysine residue causing fragmentation of Apo B (Stocker and Keaney, 2004[69]). The same process also confirms covalent adducts and an increase the negative charge of LDL molecules. Since, these sequences of events are difficult to monitor in vivo, REM and Apo B fragmentation by electrophoresis is popularly adopted in vitro protocol for assessing anti-atherogenic potential of the test compound on Cu+2 mediated oxidatively modified LDL molecules. In our study, CS treatment recorded for restoration of REM in a dose dependent manner with the highest dose (100 μg/ml) showing results comparable to that of n-LDL (Control). Also, the Ox-LDL witnessed complete disappearance of the Apo B fragment. The same made reappearance with the highest dose accounting for results comparable to n-LDL (Control). These observations strongly suggest that CS extract is efficient in preventing LDL oxidation due to rich polyphenol content that prevents oxidative damage. These credentials strengthen the claim that CS extract has the anti-atherogenic potentials.
Several studies have shown that major cell types such as the endothelial cells smooth muscle cells and monocyte derived macrophages have ability to oxidise LDL (Aviram and Rosenblat, 1994[4]; Rusinol et al., 2000[61]; Yen et al., 2008[74]). Hence, RAW 264.7 macrophages were fed with n-LDL and incubated for 24 h in presence or absence of CS extract to assess the efficacy of CS extract in preventing LDL oxidation. Decreased levels of LDL oxidation products (MDA) recorded in the present study provide testimony to the efficacy of CS extract in significantly preventing cell mediated LDL oxidation. The antioxidant property of hydro-methanolic extract of CS has already been established in our lab (Patel et al., 2012[54]) and observed effects in the present study are attributable to the efficacy of CS extract in preventing oxidative modification of LDL.
In the progression of atherosclerosis monocytes cross the endothelial barrier and differentiate into resident macrophages (Boyle, 2005[8]). The endothelial and smooth muscle cells of thoracic aorta oxidized the native LDL. This is a crucial step wherein the macrophages take up Ox-LDL via scavenger receptor (SRB1) to transform into lipid laden foam cells (Shashkin et al., 2005[64]). In our study Ox-LDL fed RAW 264.7 cells showed clear evidences of intracellular accumulation. However, visibly less number of foam cells observed in CS treated macrophages possibly indicates at the ability of CS in preventing SRB1 expression. The same needs further experimental validation.
Pathogenesis of atherosclerosis is also marked by elevated levels of intracellular oxidative stress due to formation of peroxy radicals prior to the trigger apoptotic pathway (Asmis and Begley, 2003[3]). In the present study, a peroxy radical specific stain (H2-DCFDA) produces powerful green fluorescence in macrophages that have been fed with Ox-LDL (Thounaojam et al., 2011[71]). Results obtained in our study are in accordance with other study wherein cells show high intensity of green fluorescence due to conversion of H2-DCFDA (colorless) to DCFDA (green colour) in presence of peroxy radicals. Polyphenols are free radicals scavenger and polyphenol rich CS extract used in the present study possibly scavenges peroxy radicals as evidenced by a weak fluorescence observed in Ox-LDL+CS treated group.
Metabolic performance of the cell including macrophages rest upon the metabolic normalcy of its mitochondrial (Newsholme and Newsholme, 1989[51]). Intracellular oxidative stress triggers depolarization of mitochondrial membrane and the same has been reported in Ox-LDL treated macrophages using RHO 123 stain (Huigsloot et al., 2002[34]). In the present study, RAW 264.7 cells exposed to Ox-LDL recorded a significant decrement in mitochondrial membrane potential whereas; the same was restored to normalcy in a dose dependent manner following CS treatment. These results are in accordance with the cytotoxicity assay in which the CS treated cells showed a dose dependent improvement in cell viability, possibly due to the CS extract in alleviating Ox-LDL induced mitochondrial damage.
An association between Ox-LDL to apoptosis and necrosis of foam cells has been hypothesized by several workers (Tabas, 2005[70]) and has also been demonstrated in vitro (Chang et al., 2006[13]). In the pathophysiological progression of atherosclerosis elevated levels of intracellular cholesterol play a critical role in regulation of Ox-LDL mediated apoptosis (Ryan et al., 2005[62]). In the present study, Ox-LDL treated RAW 246.7 cells recorded nuclear condensation, cell cycle arrest and apoptosis. However, CS treatment showed dose dependent increment in cell viability with more number of cells in G0 phase. CS extract has been reported to be possessing cholesterol lowering properties (Chithra and Leelamma, 1997[15]). Such efficacies of herbal extracts have discretely associated with their ability to induce reverse cholesterol transport and massive cholesterol efflux (Kaplan et al., 2001[40]). Observations recorded herein such as reduced foam cells formation and improved cell viability are attributable to hypolipidemic property of CS extract that coupled with its antioxidant potential provides multi-pronged safety to the cells. These results provide compelling evidence of anti-atherogenic potential of CS extract and hence, its efficacy was further validated in an in vivo atherogenic rodent model.
In the present study, Vit D3 + sodium Cholet + PTU and SD rat model was used to induce atherosclerosis (Cai et al., 2005[10]). In this model, PTU induced hypothyroidism and disruption of LDL receptors coupled with sodium Cholet induced increased cholesterol absorption and lower Cholesterol 7 alpha-hydroxylase activity results in hypercholesterolemia. In such circumstances vit. D3 has a detrimental effect on the structure and function of aorta, a condition that mimics in vivo atherosclerosis in humans (Cai et al., 2005[10]). Significantly elevated lipid profile of serum and aorta of atherosclerotic rats in the present study further corroborate the said hypothesis. However, lowered lipid profile following CS treatment is in agreement with the lipid lowering property of CS reported by Chithra and Leelamma (1997[15]). Serum LDL isolated from atherosclerotic rats showed elevated levels of MDA suggesting that serum LDL had undergone in vivo oxidation. However, co-supplementation of CS in atherosclerotic rats accounted for already established ability of CS in preventing LDL oxidation in an in vitro assay performed in this study.
Increased expression of cell adhesion molecules on an activated atherogenic endothelium governs and initiation and progression of atherosclerosis (Thounaojam et al., 2012[72]). VCAM-1 and p-selectin expression in aorta facilitates adhesion of leucocytes that ends in a cascade of events (Carlos and Harlan, 1994[12]; Gebuhrer et al., 1995[24]). In the present study, endothelium of thoracic aorta of CS treated atherosclerotic rats recorded significantly lowering immune localization of CAMs as capered to atherosclerotic rats. These results are in agreement with our previous report (Thounaojam et al., 2012[72]). These results further emphasized upon the anti-atherogenic potential of CS extract.
Pathological evaluation of thoracic aorta of atherosclerotic rats showed prominent atheromatous plaque formation and related damage to the vascular endothelial and smooth muscle cells. Also, calcium deposition is clearly evident. These results were greatly reduced in CS co-supplemented group and the results were comparable to that of the control group. Endothelial damage, smooth muscle cells migration, plaque formation and calcium deposition are extensively reported pathological damages in atherogenic rats. And the same are attributable to vitamin D3 injection. CS induced prevention of plaque formation; smooth muscle cell migration safe guarding of vascular endothelium and preventing calcium deposition suggest that CS extract has potential of mitigating in vivo induction of experimental atherosclerosis. These results are in synergy with previously reported therapeutic potential of CS extract and add further value to its established reputation as a medicinal dietary ingredient.
Acknowledgement
The author Dipak Patel is grateful to University Grants Commission, New Delhi for providing Financial Assistance in the form of JRFSMS scholarship.
References
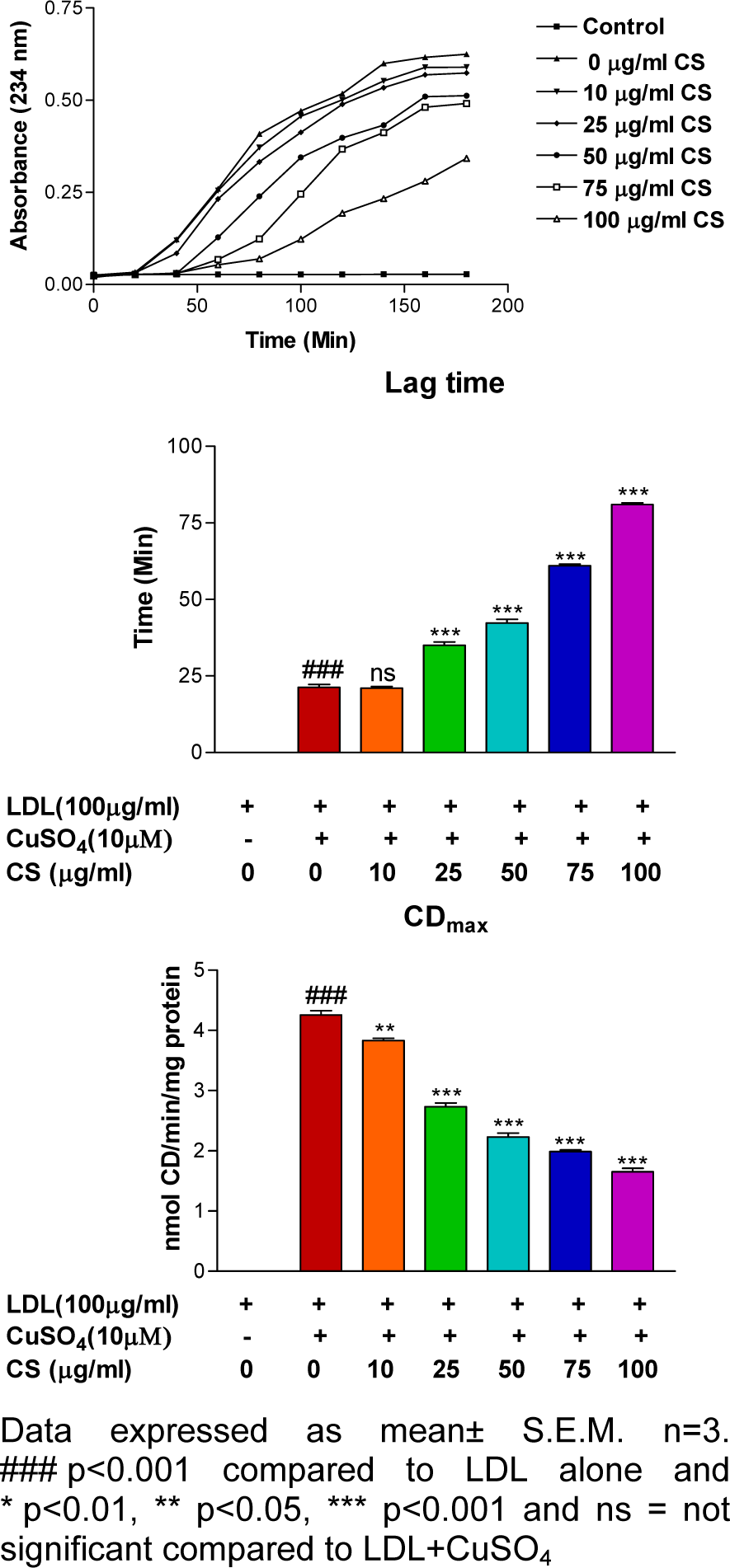
Figure 1: Effect of CS on copper-mediated LDL oxidation kinetics. Presence of CS extract accounted for a dose dependent decrement in absorbance and CDmax values and an increment in lag time
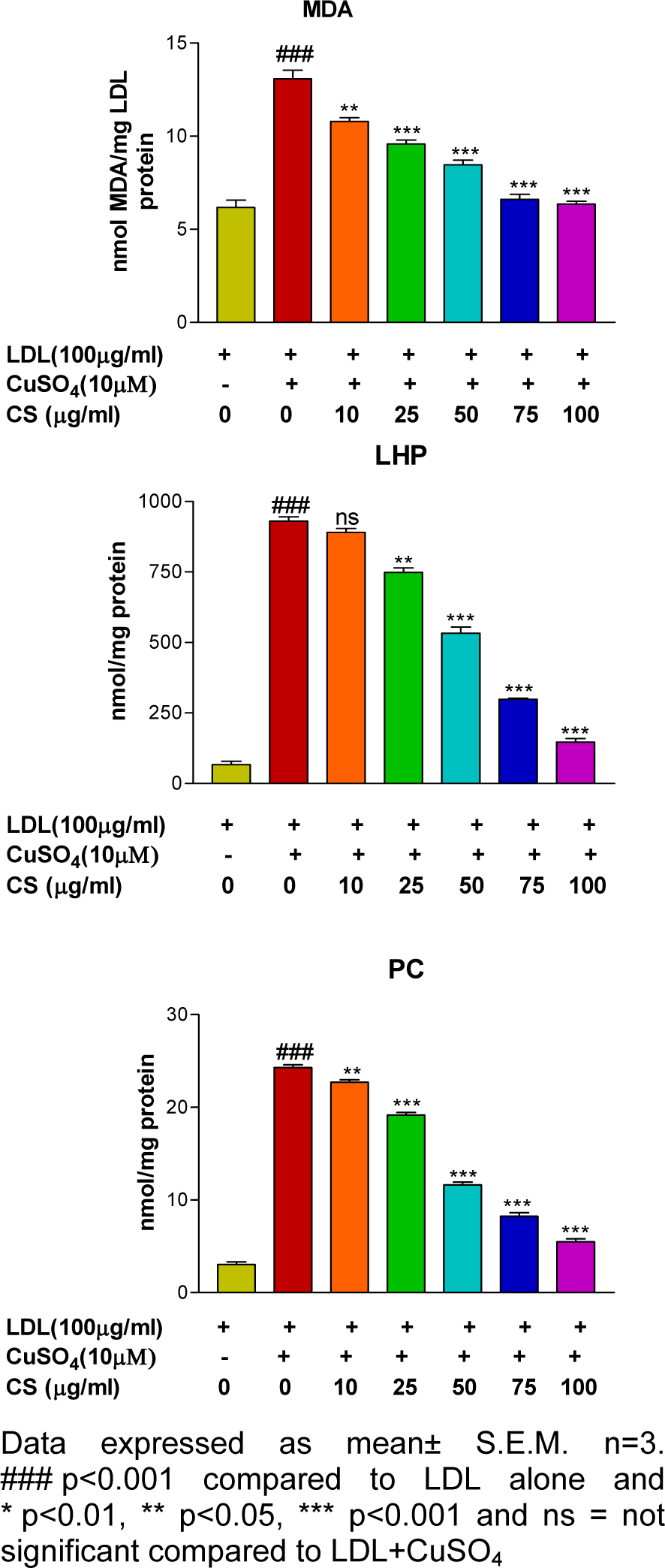
Figure 2: Effect of CS on formation of LDL oxidation products (MDA, LHP and PC) that were found to be less in presence of CS extract

Figure 3: a) Effect of CS on copper mediated ApoB fragmentation. Arrow indicates the ApoB band (L1) that disappears following LDL oxidation (L2) but reappears in presence of CS (L3-L7). b) Relative electrophoretic mobility. Arrow indicates mobility of nLDL (L1) that increases following oxidative modification (L2) but gets restored in presence of CS (L3-L4)
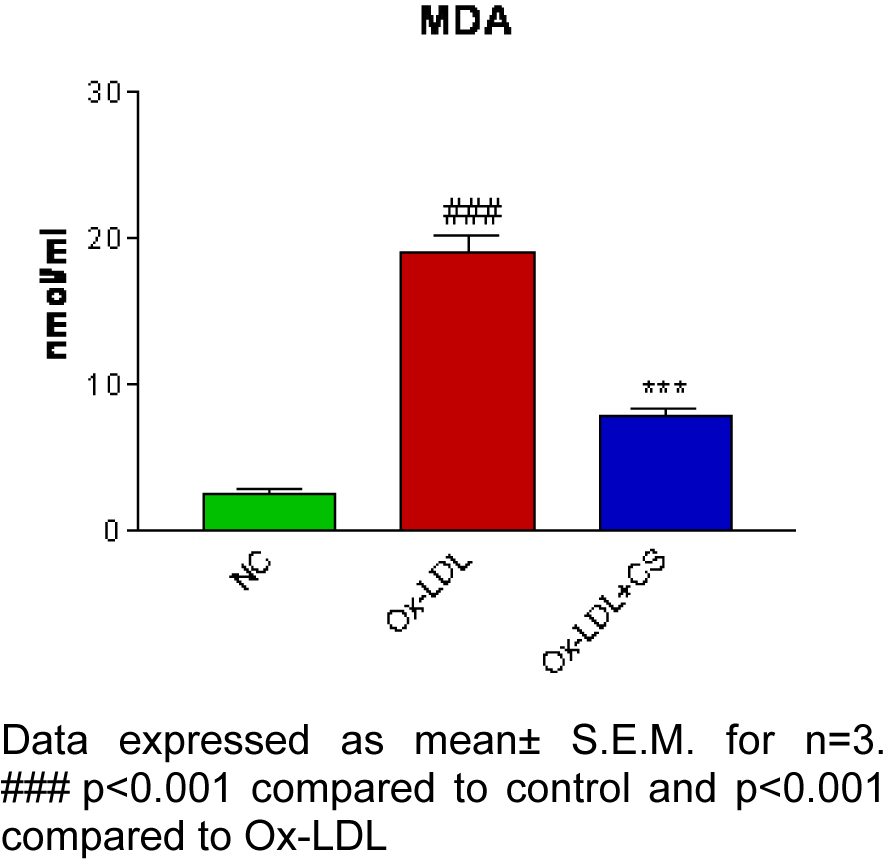
Figure 4: Effect of CS on RAW 264.7 cell mediated LDL oxidation. Less amount of MDA formed in presence of CS
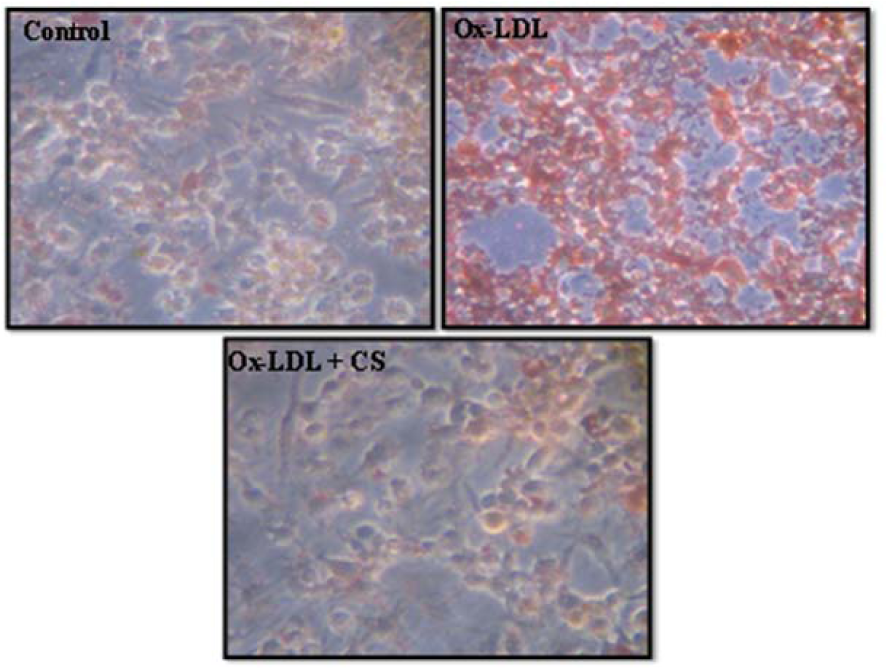
Figure 5: Foam cell formation assay in RAW 264.7 cells. Control (untreated), Ox-LDL: Cells treated with Ox-LDL and Ox-LDL+CS: treated with Ox-LDL in presence of 100 μg/ml CS extract. Red cells indicate Oil red O positive cells due to intracellular accumulation of cholesterol
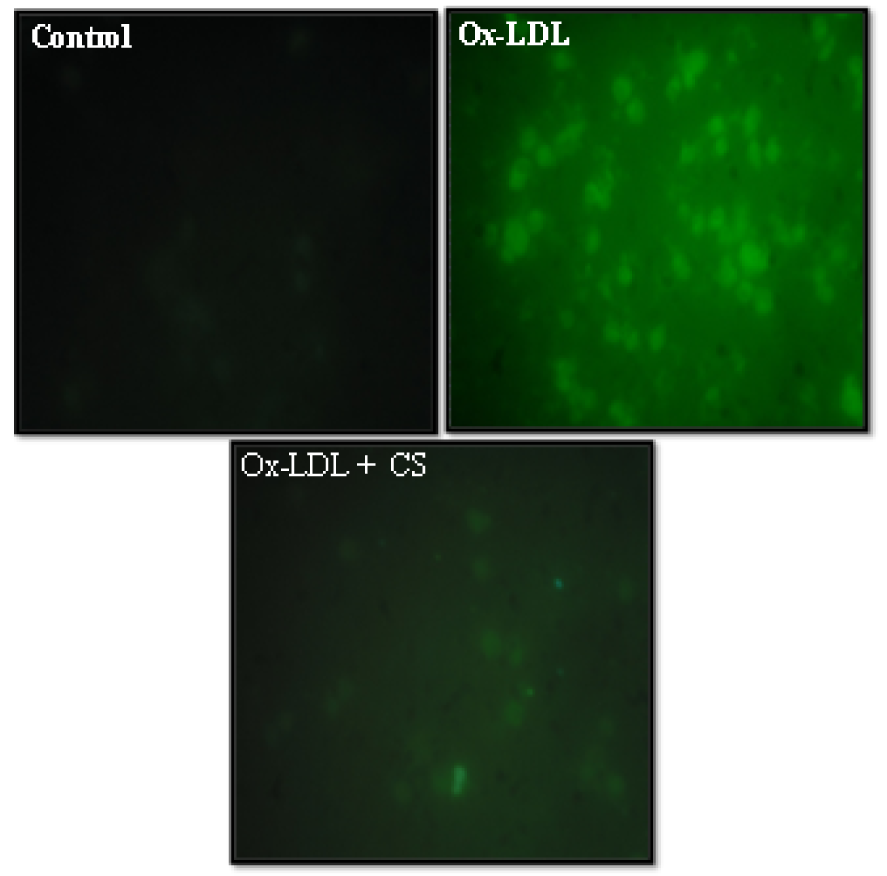
Figure 6: Peroxyl radical generation (DCF-DA staining) of RAW 264.7 cells: Control (untreated), Ox-LDL: Cells treated with Ox-LDL and Ox-LDL+CS: treated with Ox-LDL in presence of 100 μg/ml CS extract. Fluorescent intensity indicates intracellular oxidative stress
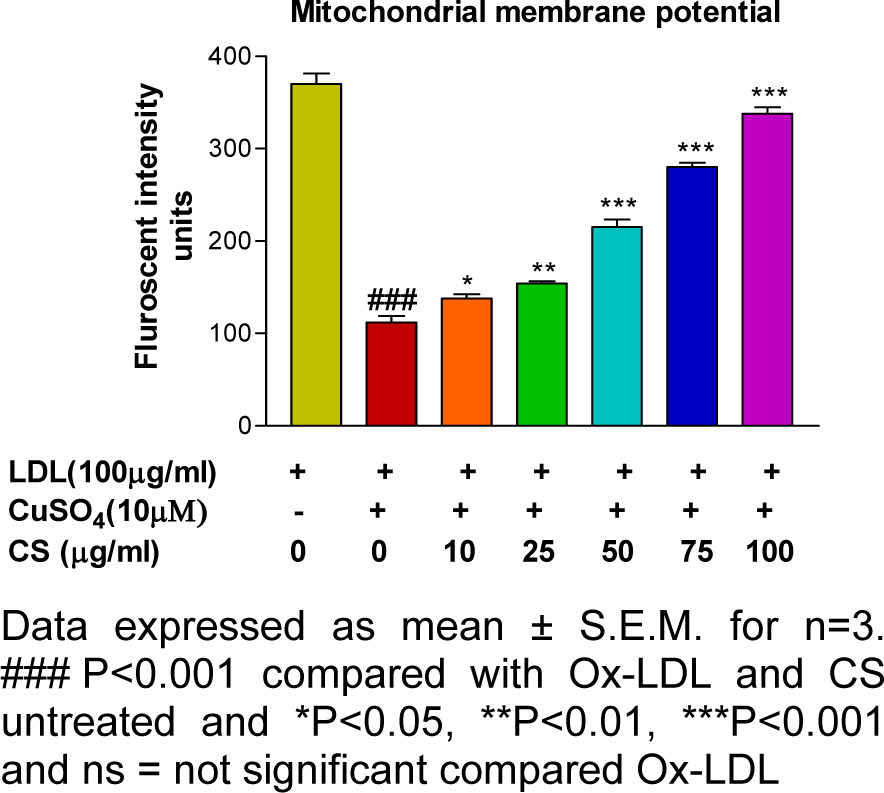
Figure 7: Mitochondrial membrane potential (Rhodamine 123 staining) of RAW 264.7 cells. Ox-LDL treatment accounts for decrement in fluorescent intensity units
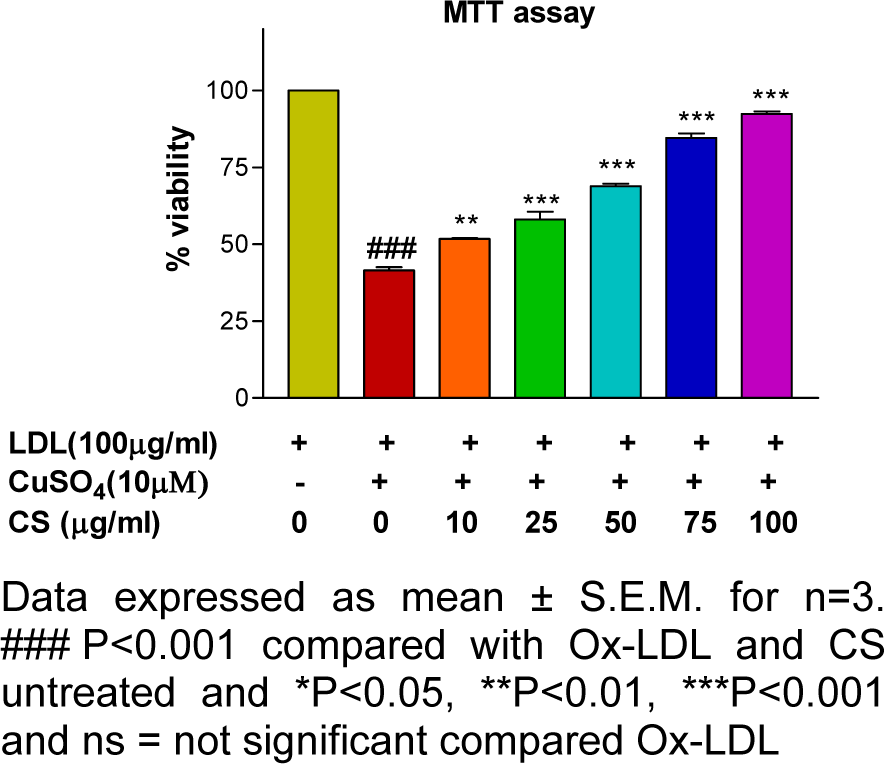
Figure 8: Cell viability (MTT) assay: Ox-LDL accounted for decrement in cell viability that was alleviated in presence of CS extract
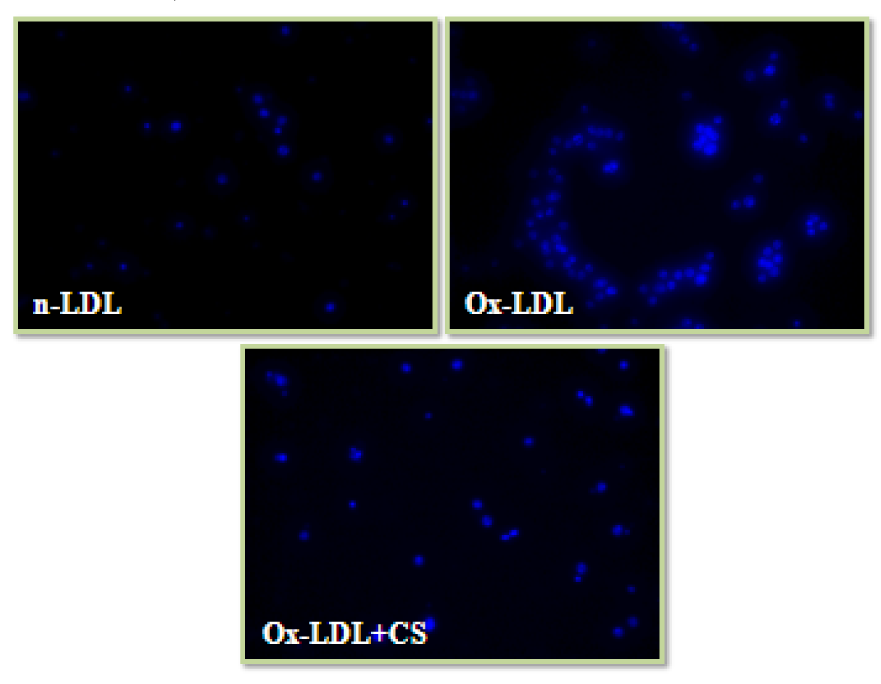
Figure 9: Nuclear condensation (DAPI staining) of RAW 264.7 cells: Control cells, Ox-LDL: Cells treated with Ox-LDL and Ox-LDL+CS: treated with Ox-LDL in presence of 100 μg/ml CS extract
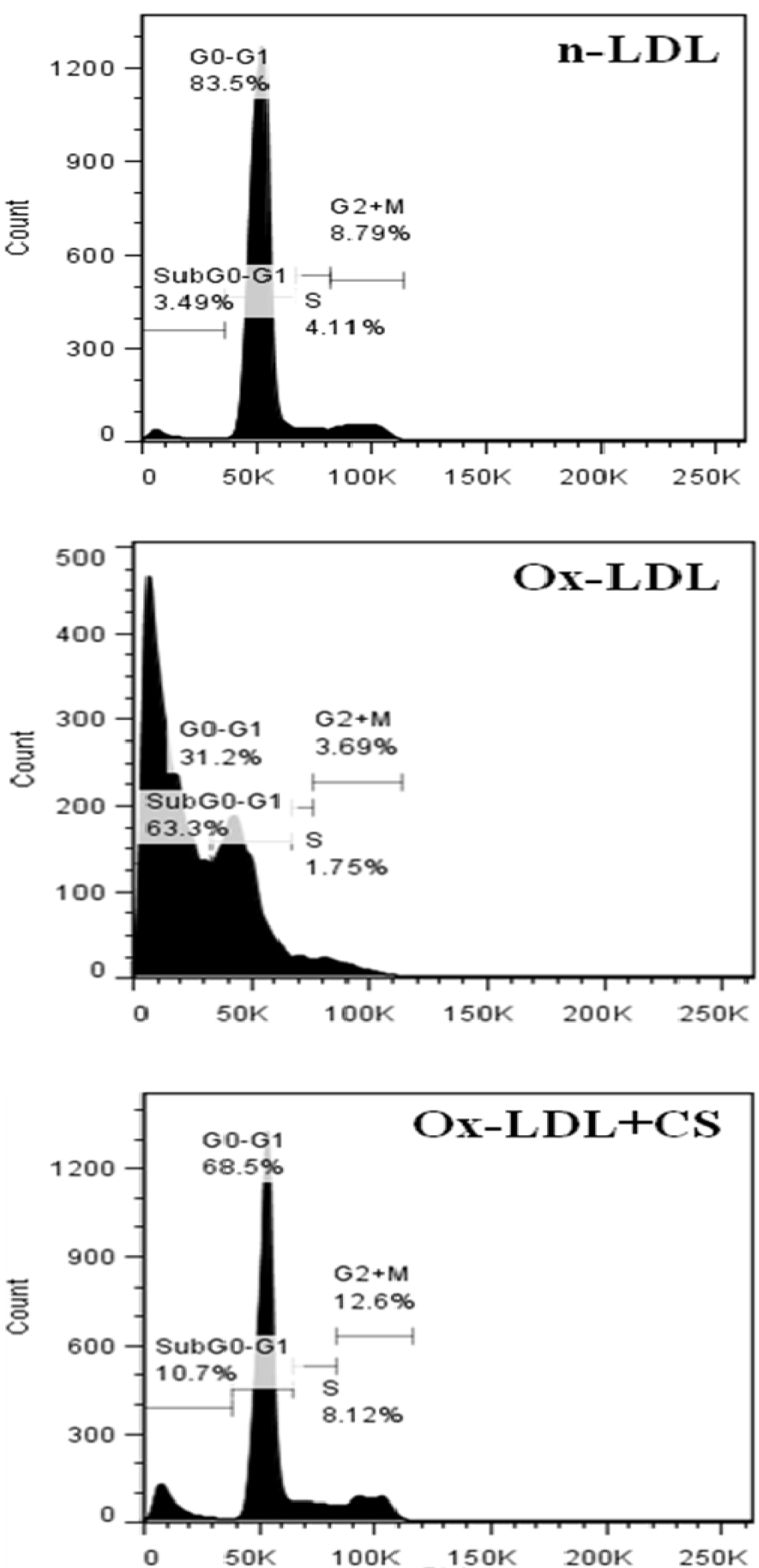
Figure 10: Cell cycle analysis of RAW 264.7 cells. Control cells, Ox-LDL: Cells treated with Ox-LDL and Ox-LDL+CS: treated with Ox-LDL in presence of 100 μg/ml CS extract. More number of cells were present in sub G0 phase following Ox-LDL treatment
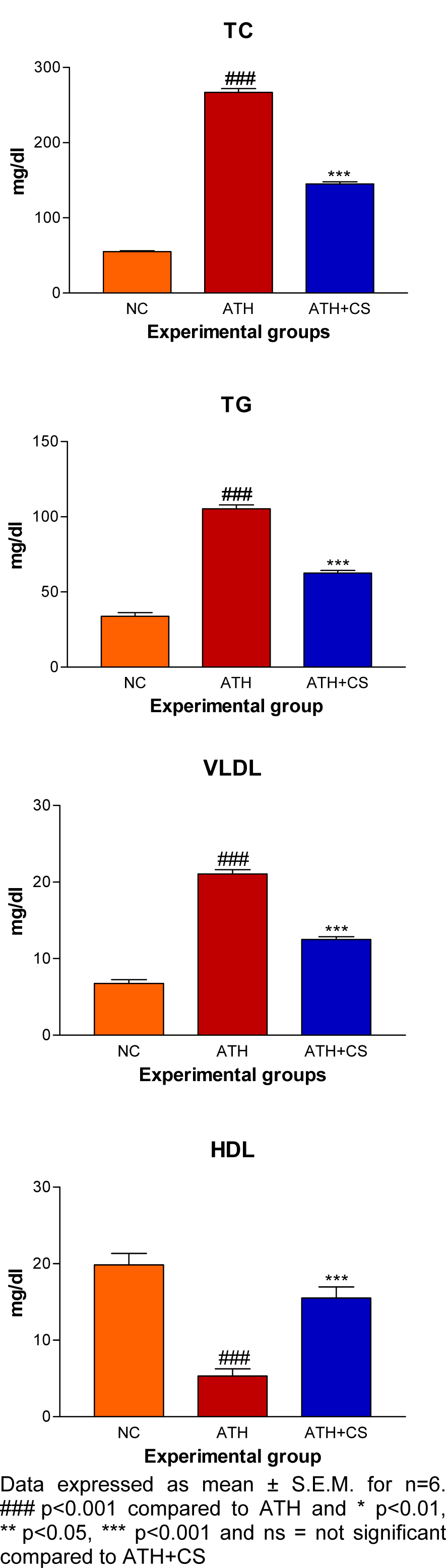
Figure 11: Effect of CS on lipid and lipoprotein profiles of control (NC), atherogenic diet fed (ATH) and ATH+CS supplemented rats
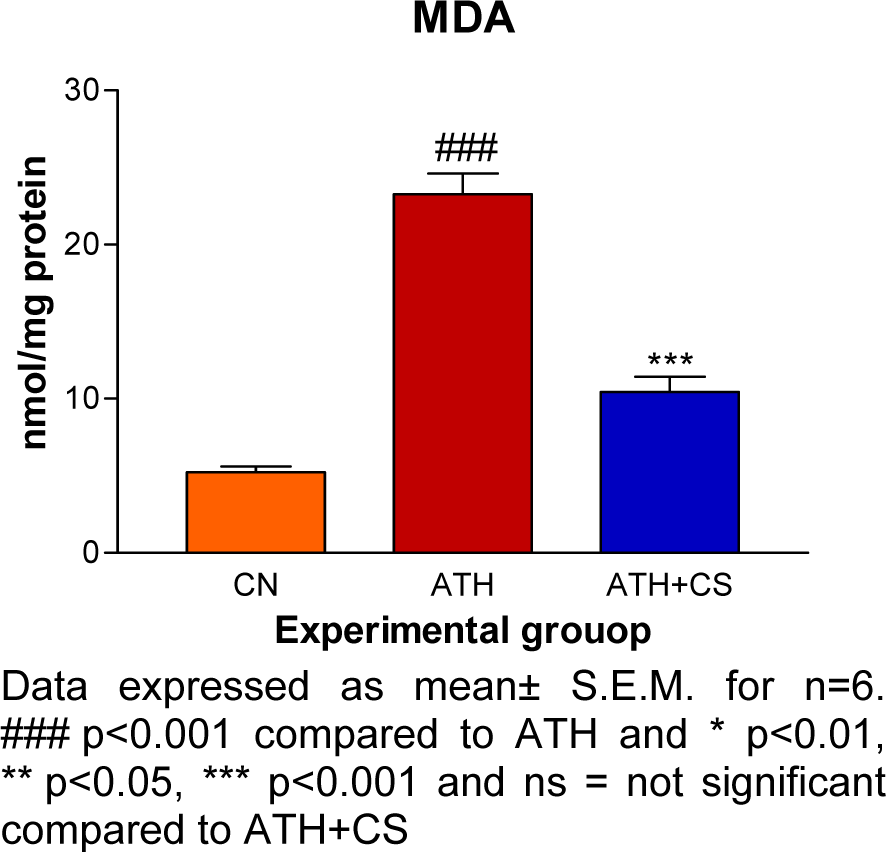
Figure 12: Effect of CS on MDA level in control (NC), atherogenic diet fed (ATH) and ATH+CS supplemented rats
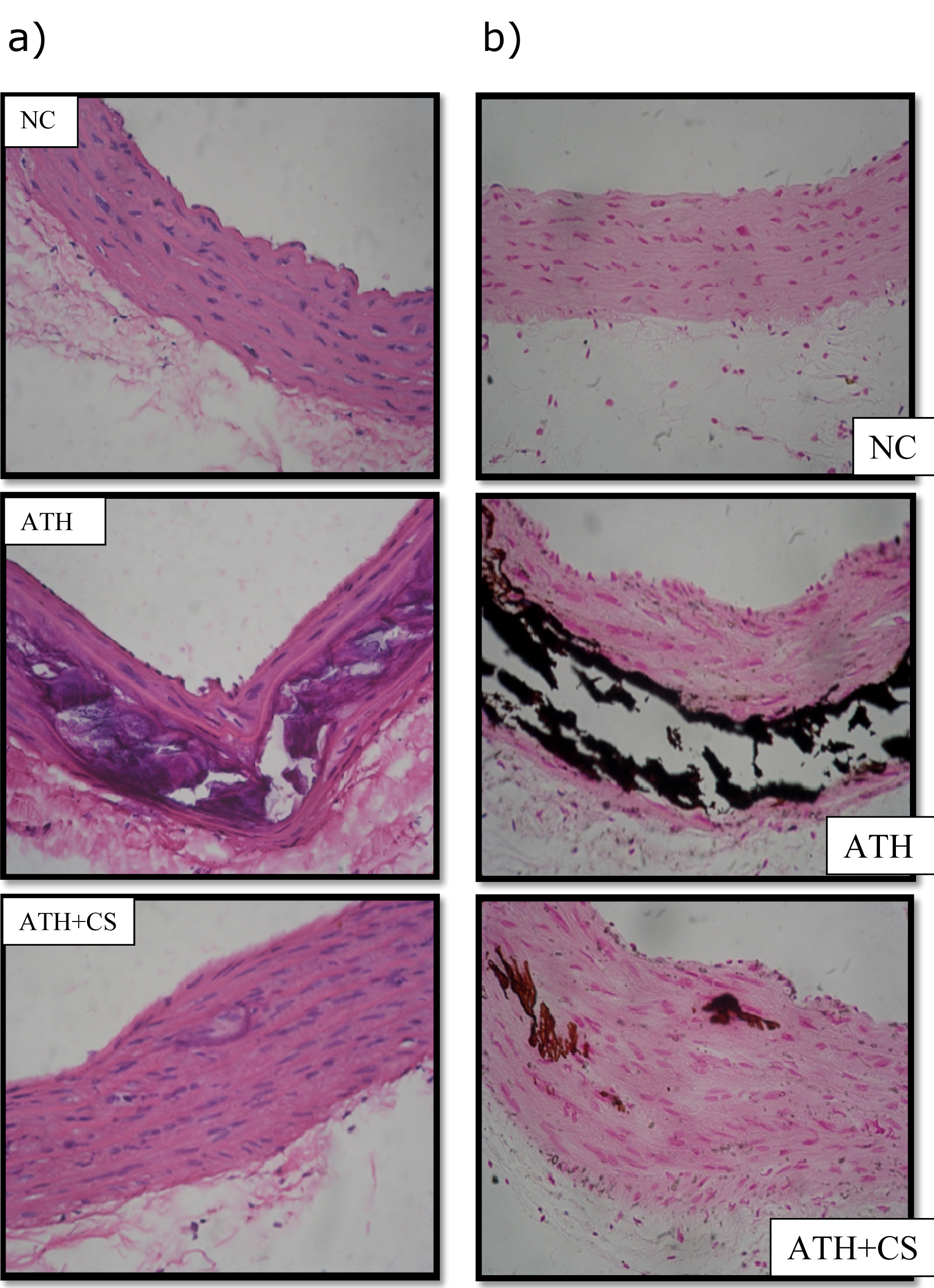
Figure 13: a) Histopathology of thoracic aorta (HxE stained) control (NC), atherogenic diet fed (ATH) and ATH+CS supplemented rats. Note the atheromatous plaque formation following ATH treatment. b) Calcification (von Kossa stain) of thoracic aorta of control (NC), atherogenic diet fed (ATH) and ATH+CS supplemented rats
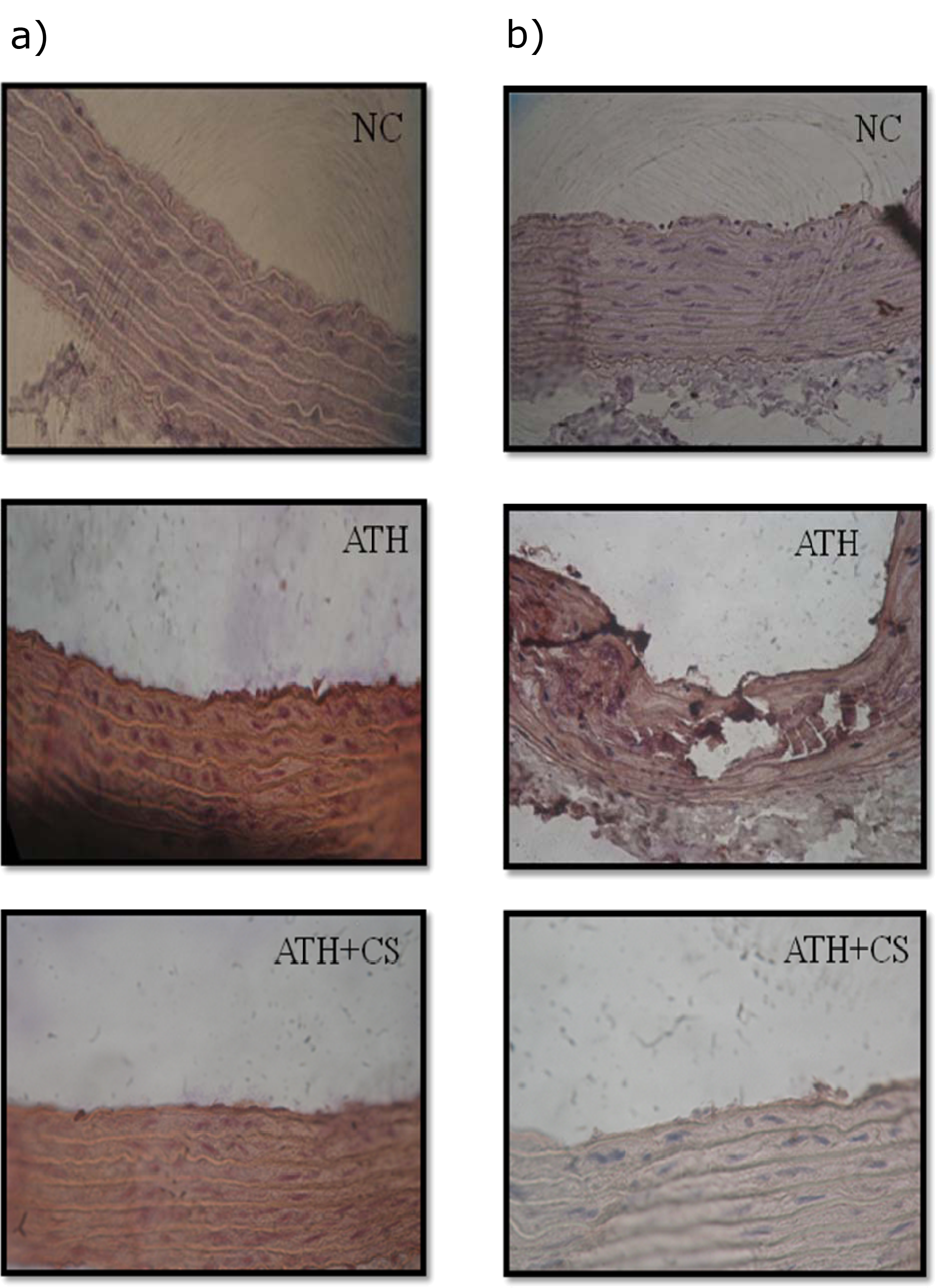
Figure 14: a) Immunohistochemical localization of VCAM1 in thoracic aorta of control (NC), atherogenic diet fed (ATH) and ATH+CS supplemented rats. b) Immunohistochemical localization of p-selectin in thoracic aorta of control (NC), atherogenic diet fed (ATH) and ATH+CS supplemented rats
[*] Corresponding Author:
Ranjitsinh Devkar, Division of Phytotherapeutics and Metabolic Endocrinology, Department of Zoology, Faculty of Science, The M. S. University of Baroda, Gujarat, India; Tel. No.: +91-9825935445, Fax No: +91-0265-2342109, eMail: phyto_met@yahoo.com