Research article
Novel inhibitor discovery against aromatase through virtual screening and molecular dynamic simulation: a computational approach in drug design
Sako Mirzaie1, Latifeh Chupani2, Ebrahim Barzegari Asadabadi2, Ahmad Reza Shahverdi3, Mostafa Jamalan2[*]
1Department of Biochemistry, Sanandaj Branch, Islamic Azad University, Sanandaj, Iran2Department of Biology, Faculty of Science, Science and Research Branch, Islamic Azad University, Tehran, Iran
3Department of Pharmaceutical Biotechnology, Faculty of Pharmacy and Biotechnology Research Center, Tehran University of Medical Sciences, Tehran, Iran
EXCLI J 2013;12:Doc168
Abstract
Inhibition of aromatase (CYTP450) as a key enzyme in the estrogen biosynthesis could result in regression of estrogen-dependent tumors and even preventing the promotion of breast cancer. Although today potent steroid and non-steroid inhibitors of aromatase are available, isoflavanone derivatives as natural compounds with least side effects have been described as the candidate for a new generation of aromatase inhibitors. 2a as an isoflavanone derivative is the most potent inhibitor of aromatase, synthesized by Bonfield et al. (2012[7]). In our computational study, the mentioned compound was used as the template for virtual screening. Between 286 selected compounds with 70 % of structural similarity to 2a, 150 of them showed lower docking energy in comparison with 2a. Compound 2a_1 with 11.2 kcal/mol had the lowest docking energy. Interaction of 2a_1 with aromatase was further investigated and compared with 2a and androstenedione (ASD) as a natural substrate of aromatase, through 20 ns of molecular dynamic simulation. Analysis of trajectories showed, while ASD interacts with aromatase through hydrogen bonds and 2a just interacts via hydrophobic forces, 2a_1 not only accommodates in the hydrophobic active site of aromatase in a suitable manner but it also makes a stable coordination with iron atom of aromatase heme group via OB.
Keywords: aromatase, androstenedione, aromatase inhibitor, structural-based virtual screening
Introduction
Breast cancer is the first widespread kind of cancer among women and after lung cancer is the major cause of death due to malignancy (Hernandez et al., 2010[19]). Estrogens, through various mechanisms, have the predominant role in induction and proliferation of breast cancers (Santen and Harvey, 1999[43]). Estrogens could cause carcinogenesis synergically through accelerating cell division, which results in abridging the time of DNA repairing phase, and as additive fashion, with creation of quinines via their metabolic pathway (Santen et al., 1999[45]). Breast cancer tumors can be categorized as hormone-depended or independent of hormone stimulation subtypes (Santen and Harvey, 1999[43]). In hormone-dependent case of breast cancer, estrogen plays a crucial role in development of tumors through binding to estrogen receptors, which leads to induction of intracellular signaling cascades (Berstein et al., 1999[5]; Buzdar, 1999[8]). Estrogen-dependent carcinomas were reported in near 60 percent of premenopausal and 75 percent of post-menopausal breast cancer patients (Chen, 1998[11]). So, inhibition of aromatase as the rate limiting enzyme in the estrogen biosynthesis pathway could limit the growth and development of estrogen-dependent tumors. In addition, such inhibition might cause an attenuation of estradiol level in breast tissue, which finally results in prevention of the tumor promotion process (Santen et al., 1999[45]).
Aromatase, also known as Cytochrome P450, is a key enzyme which converts cholesterol to estradiol E2 as the most biologically active estrogen (Simpson et al., 1994[47]). Aromatase contains a catalytic heme group with an iron atom forming an octahedral coordination geometry which is responsible for oxidation of substrate (Favia et al., 2006[15]). The iron atom in heme group of aromatase is coordinated by four nitrogen atoms of protoporphyrin IX and a thiolate group from Cys437. Its sixth coordination is occupied by water molecule which is necessary for hydroxylation process (Auclair et al., 2001[3]; Meunier et al., 2004[37]). In the presence of three moles of oxygen and three moles of NADPH, aromatase catalyzes the transformation of one mole of androstenedione (ASD) to estrone E1 through a three-step reaction which is the rate limiting step in the synthesis of estradiol E2 (Meunier et al., 2004[37]). Synthesis of 19-hydroxyandrostenedione from ASD through hydroxylation of C19, production of 19-gem-dihydroxyandrostenedione via another hydroxylation on C19 and generation of estrone E1 which accompany with extrication of a water molecule and production of a formate molecule, are the three steps of an aromatization process which is mediated by aromatase (Meunier et al., 2004[37]). Moreover, aromatase has a crucial role in metabolisms of xenobiotics, drugs and other kinds of steroids (Guengerich, 2004[17]; Williams et al., 2000[51]).
Aminoglutethimide and testololactone are the first-generation inhibitors of aromatase which result in regression of tumors in administrated patients. They don't act as potent kinds of inhibitors and their unspecific action leads to induction of side-effects (Santen et al., 1990[44]). Today, FDA-approved drugs such as exemestane (Coombes et al., 2004[14]) as an steroid inhibitor, letrozole (Coates et al., 2007[13]) and anastrozole (Locker et al., 2007[35]) as non-steroid specific aromatase inhibitors (AIs) can efficiently decrease the recurrence rate of hormone-dependent breast cancer. Exemestane is a mechanism-based inhibitor and inhibits aromatase in an irreversible manner (Hong et al., 2007[21]) but other AIs like letrozole and anastrozole act in a competitive and reversible inhibition procedure, so their mechanism of action still remains unclear (Hong et al., 2011[20]; Kao et al., 1996[27]).
Although today, non-steroid aromatase inhibitors (NSAIs) are the most progressive kinds (Bhatnagar, 2007[6]), the occurrence probability of side-effects due to long-term administration has motivated new attempts for development of a new generation of AIs based on natural compounds and their derivatives such as coumarin, lignin and flavonoids (Sakamoto et al., 2010[41]). Structural modification of isoflavanone through various approaches by Bonfield et al. (2012[7]) resulted in production of new derivatives with greater ability for inhibition of aromatase activity. Among synthesized derivatives, the compound named 2a (Figure 1B(Fig. 1), Bonfield et al., 2012[7]) with IC50 of 0.26 µM was the most efficient inhibitor of aromatase (Bonfield et al., 2012[7]). In the present study, inhibitory mechanism of compound 2a was further investigated and its interaction with aromatase in the presence of its natural substrate, ASD, was studied through 20 ns of molecular dynamic simulation (MD simulation). Furthermore, structure-based virtual screening method was applied to develop new compounds with more potent inhibitory effect.
Materials and Methods
Ligand-based virtual screening
The ZINC database (Irwin et al., 2012[25]), a free virtual screening database containing over 21 million compounds in ready-to-dock 3D formats, was investigated for structures with 70 % similarity to 2a (http://zinc.docking.org/). 286 purchasable compounds with 70 % of structural similarity to 2a were captured in this way and used for structure-based virtual screening.
Biological activity
The negative logarithm of the measured IC50 (M) against human aromatase (pIC50) (Bonfield et al., 2012[7]) was used in Pearson correlation.
Structure-based virtual screening
All of the 286 captured compounds were docked against the crystal structure of aromatase (PDB code: 3S79) obtained from protein databank (Berman et al., 2000[4]). Acquired file contains aromatase with its natural substrate, ASD, in 2.75 Å resolution. AutoDock Vina was used as docking tool (Trott and Olson, 2010[49]) and AUDocker LE was applied for automatic consequential docking process (Sandeep et al., 2011[42]). All of default settings were assigned during the docking procedure. Usually, docking programs use a scoring function to approximate the standard chemical potentials of the defined complex system. Scoring function of AutoDock Vina is defined as below (Trott and Olson, 2010[49]):
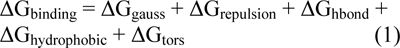
The scoring function mentioned above was calibrated with approximately 1300 receptor-ligand complexes. Furthermore, its algorithm search is gradient-based local search (Chang et al., 2010[10]). The conformation-dependent part of the scoring function AutoDock Vina could be shown as equation 2:

which ftitj is the interaction function of two atoms i and j. rij is the distance between atoms i and j (Trott and Olson, 2010[49]). Value c is calculated for both intra and inter-molecular bonds:

In order to linking between windows system and AutoDock Vina in our virtual screening study, AUDocker LE software was used. By the aid of this software, the ligand efficiency could also be calculated. By dividing the theoretical ΔGbinding value by the number of non-hydrogen atoms, the ligand efficiency value is achieved (Abad-Zapatero and Metz, 2005[1]; Hopkins et al., 2004[22]):

where ΔG is proportional to Kd :

and N is the number of non-hydrogen atoms.
Molecular simulation of aromatase and aromatase-ligand complexes
Interaction of ASD, 2a and 2a_1 which had the lower docking energy between all of the docked compounds were more investigated through 20 ns of MD simulation. With regard to this note that the structure of protein determined by X-ray crystallography is lacked of hydrogen, at the first step, missing hydrogen atoms were added to aromatase PDB file. The crystal structure of aromatase was prepared for MD simulation through mild minimization, and then solvation within a cubic shaped water cage with 1 Å of distance between protein periphery and the cube edges. System neutralization was applied by addition of three chloride ions and then, another energy minimization step was implemented. All MD simulations were carried out by the GROMACS 4.5.5 package using the ffgmx force field (Gromos87) (van der Spoel et al., 2005[50]). Before the production MD simulation run, the temperature of crystal structure was reached to 300 K and equilibrated during 50 ps in NVT ensemble. After another equilibration step which performed through 100 ps of NPT simulation, the crystal structure of aromatase was simulated through 20 ns of NPT ensemble. Molecular dynamic simulations of aromatase alone and in the presence of ASD, 2a and 2a_1 were individually done as mentioned above. Topologies and coordinations of ligands were generated by PRODRG web server (Schuttelkopf and van Aalten, 2004[46]). For determination of hydrogen bonds between aromatase with various ligands, default values of Gromacs were assigned. Based on default, r is the distance between heavy atom which is incorporated in generation of hydrogen bond in such a way that r ≤ 0.35 nm. 3D-images of aromatase or its complex with docked ligands were rendered by UCSF Chimera (Goddard et al., 2005[16]) or Pymol (Humphrey et al., 1996[23]). 2D-images of aromatase-ligand interactions were created by LigPlot (Laskowski and Swindells, 2011[34]).
Results and Discussion
Internal validation phase
Based on the free energy of binding of aromatase inhibitors, the more active and potent candidates were selected for further studies. So, its necessary to evaluate the accuracy of the calculated free energy of binding. For this purpose, eleven proven inhibitors were taken from the literature (Bonfield et al., 2012[7]) and assigned as a test set. Table 1(Tab. 1) (Bonfield et al., 2012[7]) shows the compound structures with their pIC50 and predicted free energy of binding (Bonfield et al., 2012[7]). Figure 2(Fig. 2) shows the correlation between the experimental IC50 and the estimated free energy of binding of test set which was calculated by Pearson correlation.
The correlation between two variables reflects the degree to which the variables are related. The most common measure of correlation is the Pearson Product Moment Correlation (called Pearson's correlation for short). Pearson's correlation reflects the degree of linear relationship between two variables. It ranges from +1 to -1. A correlation of +1 means that there is a perfect positive linear relationship between variables. Correlation value between experimental and theoretical data described as Pearson correlation coefficient or R2. The calculated R2 for this simulation was 0.88 which showed an acceptable correlation. This step is too important because the screening phase of this study is based on docking energy of each compound. It should be noted that the molecular docking study in validation phase was done against the intact crystal structure of aromatase.
Virtual screening through docking energy
Among 286 compounds with structural similarity to 1a (Figure 1(Fig. 1)), which were docked against the minimized structure of aromatase, 150 compounds showed lower docking energy, compared with 2a. The docking energy for 2a was -8.5 kcal/mol, while 22 screened compounds had docking energies below -10 kcal/mol. The ASD as a natural substrate of aromatase by -13.8 had the lowest level of docking energy among all of docked compounds. Energy of docking for 21 compounds with 70 % of structural similarity to 2a, which had docking energy below -10 kcal/mol is indicated in Table 2(Tab. 2) and their chemical structures are shown in Figure 3(Fig. 3).
On validation phase of docking process, ASD which is completely docked to aromatase was imposed on ASD coordination in available crystal structure. RMSD of docking for ASD in comparison with its coordination in the crystal structure was zero. ASD in aromatase-ASD crystal structure is located near the heme group. This compound interacts with Met374 just via one hydrogen bond which is generated through O2 (Figure 1(Fig. 1)), the oxygen atom attached to cyclohexane ring of ASD through double bond. Moreover, 2a and 2a_1 were located near ASD coordination in aromatase crystal structure through docking (Figure 4(Fig. 4)). Interactions of ASD, 2a and 2a_1 with aromatase were further investigated through 20 ns of MD simulation.
Interaction of ASD with aromatase through 20 ns of MD simulation
ASD is the natural substrate of aromatase and has a high affinity for it, as can be seen from its docking energy. Root mean square deviation (RMSD) values for aromatase as alone and in complex with ASD are different through 20 ns of MD simulation (Figure 5(Fig. 5)). Binding of ASD to aromatase after 6 ns could cause structural alternation, which could be seen from the distinct RMSD profiles. This different conformation for aromatase in the presence of ASD was reported previously by Hong et al. (2007[21]). They showed that proteolytic digestion of aromatase by the trypsin, without the substrate or in the presence of ASD, and even in the presence of reversible inhibitors results in various profiles of aromatase fragments on SDS-PAGE (Hong et al., 2007[21]). ASD, through an extended network of hydrogen bonds (H-bonds) which remains stable through 20 ns of MD simulation, has a high affinity for active site of aromatase. The average number of hydrogen bonds between ASD and aromatase during the simulation time is 3.21 (Figure 6(Fig. 6)). O1 of ASD makes a stable hydrogen bond with Thr310. Its O2 atom also interacts with Met374 and Arg115 of aromatase through H-bonds (Figure 7(Fig. 7)). The role of Thr310 in aromatase active site as a participative residue was previously emphasized by site-directed mutagenesis (Chen and Zhou, 1992[12]). T310S and T310C mutated types of aromatase just showed 48.8 % and 2.4 % of wild-type Vmax, respectively (Chen and Zhou, 1992[12]). The length of hydrogen bond generated between Thr310:HG1 and O1 of aromatase during 20 ns of MD simulation is 2.301 Å. So, increase in H-bond length by replacement of threonine by serine with a shorter side chain, leads to the attenuation of H-bond and decreasing the Vmax of enzyme (Chen and Zhou, 1992[12]). Replacement of Thr310 with a cysteine residue, disrupts this H-bond and causes aromatase to become nearly inactive (Chen and Zhou, 1992[12]). Root mean square fluctuation (RMSF) of alpha carbons for Arg115 and Thr310 are also different between aromatase as free of substrate and in aromatase-ASD complex through 20 ns of MD simulation (Figure 8(Fig. 8)).
2a as isoflavanone derivative interacts with aromatase through hydrophobic interactions
2a as the most potent aromatase inhibitor, which is synthesized by Bonfield et al. (2012[7]), interacts with aromatase through hydrophobic interactions (Figure 9A(Fig. 9)). The average number of hydrogen bonds, which are formed between 2a and aromatase through 20 ns is 0.019 (Figure 6(Fig. 6)). Based on Figure 7A(Fig. 7), it seems that hydrophobic interactions are the most contributing forces in the stability of aromatase-2a complex. The hydrophobic pocket formed by residues like Ile133, Phe134, Ala306, Thr310, Leu372 and Leu477 surrounds 2a. In fact, the RMSF values, which are reported here for alpha carbons of these residues through 20 ns of MD simulation in the presence of 2a may arise from their hydrophobic interaction with 2a (Figure 9B(Fig. 9)). The coordination of O2 of 2a with iron atom of aromatase heme group is so volatile. The average distance between O2 with Fe atom from the heme group is 0.632 nm, which seems too long to result in formation of a stable coordination (Figure 10(Fig. 10)).
Interaction of 2a_1 with aromatase via hydrophobic interactions and oxygen-iron coordination
Although 2a_1 (3-[(2,3,5,6-tetramethylphenyl)-carbonyl]-2,3-dihydro-1-benzofuran) does not interact with aromatase through hydrogen bonds (Figure 6(Fig. 6)), it extensively interacts with aromatase via hydrophobic interactions and furthermore makes a stable oxygen-iron coordination with the heme group of aromatase. 2a_1 is properly located in the hydrophobic active site of aromatase near its heme group. Tetramethylphenyl moiety of 2a_1 is clamped by the hydrophobic side chain of Trp224, Val373 and Met374 and its benzofuran moiety interacts with Phe134 from aromatase (Figure 11A(Fig. 11)). The presence of 2a_1 in the active site of aromatase affects RMSD of its heme group through 20 ns of the simulation process (Figure 5(Fig. 5)). Average RMSD for heme group of the ligand-free aromatase is 0.136 nm, while in the presence of 2a_1 the average RMSD of heme is 0.072 nm during 20 ns of MD simulation. 2a_1 interacts with heme group of aromatase through its O2 atom which makes a stable coordination throughout the simulation time. In the ligand-free form of aromatase, six coordinations of Fe atom from the heme group are occupied by four nitrogen atoms from heme group, a thiolate from Cys437 and an oxygen atom from the water molecule which is used for aromatization of ASD. In the presence of 2a_1, Fe2+ atom of heme could make a stable coordination with O2 atom from 2a_1. At the beginning of the simulation process, O2 atom of 2a_1 is located in the 0.43 nm of iron atom from heme group, but after just 0.2 ns of MD simulation it reaches to 0.15 nm of iron atom and remains in that position during the remaining time of simulation. The average distance between O2 from 2a_1 and Fe atom is 0.173 nm for 20 ns of MD simulation (Figure 10(Fig. 10)).
Conclusion
Phytoestrogens are a natural class of compounds, which are found in human dietary like grapes, soybean and berries (Adlercreutz, 1995[2]; Mazur et al., 2000[36]; Kiely et al., 2003[33]). Flavonoids subclasses like flavanones, flavones and isoflavonoids and lignans compose two main groups of phytoestrogens (Karkola and Wahala, 2009[29]; Milder et al., 2004[38]). Size, shape and charges of some kinds of phytoestrogens are similar to those of estrogen and so have a great potential to be used as aromatase inhibitors (Ibrahim and Abul-Hajj, 1990[24]; Jeong et al., 1999[26]; Kellis and Vickery, 1984[32]; Kao et al., 1998[28]). Role of these compounds in reducing occurrences of breast cancer has previously been emphasized (Adlercreutz, 1995[2]). Many of the potent inhibitors of aromatase which are used today as efficient drugs, like letrozole or triazole make a stable coordination through their nitrogen atoms with iron atom of aromatase heme group (Miller et al., 2008[40]; Hamilton and Volm, 2001[18]). It seems that flavonoids not only could accommodate in the hydrophobic active site of aromatase through their hydrophobic moieties, but they also could make a stable coordination with Fe atom of the heme group through their oxygen atom (Kellis and Vickery, 1984[32]; Kellis et al., 1986[31], 1987[30]). Through procedures like microwave-assisted, gold-catalyzed annulations of reaction of hydroxyaldehyde and alkynes on A and B rings of isoflavanones, Bonfield and his coworkers made a series of compounds among which 2a (6-methoxy-3-phenylchroman-4-one) is the most potent aromatase inhibitor with the IC50 value of 0.26 µM (Bonfield et al., 2012[7]). In spite of all advantages of natural compounds like isoflavanone derivatives, their inhibitory effect on aromatase is so weak in comparison with available aromatase inhibitors like anastrozole with IC50 of 12 nM (Miller, 1999[39]). In current study, chemical structure of 2a was used as a template for virtual screening through docking against available crystal structure of aromatase. Among 286 compounds with 70 % of structural similarity to 2a, 3-[(2,3,5,6-tetramethylphenyl)carbonyl] -2,3-dihydro-1-benzofuran, which was named 2a_1, showed the highest affinity for aromatase. While the energy of docking for 2a were -8.5 kcal/mol, docking energy for 2a_1 was -11.2 kcal/mol. Interactions of 2a_1 with aromatase was more investigated through 20 ns of MD simulation and compared with ASD and 2a interactions. Hydrogen bonds are the main forces through which ASD interacts with aromatase. MD simulation studies showed that ASD extensively interacts with aromatase via a stable network of hydrogen bonds with Met374, Arg115 and Thr310 throughout the time of simulation. 2a as the most important inhibitor against aromatase which was synthesized by Bonfield et al. (2012[7]) just accommodates suitably in the hydrophobic active site of aromatase but could not coordinate with Fe atom of the heme group via its oxygens. Role of the hydrophobic residues like Ala306, Thr310, Trp224, IIe133, Phe134, Val370, Leu372, Val373, Met374 and Ser478 in the active site of aromatase has been previously proven (Yadav et al., 2011[52]). In fact, replacement of carbonyl groups with more hydrophilic groups in the aromatase inhibitors leads to destabilization of the enzyme-compound interaction, decreasing its inhibitory activity (Cepa et al., 2008[9]). In addition, another study has reported that hydrophobic amino acids such as Ile132, Ile133, Ile305, Phe148, Met303, and Ala306 may play a critical role in the binding to the active site (Takahashi et al., 2010[48]). Our study demonstrated that 2a_1, which was selected through the virtual screening process, not only interacts suitably with the hydrophobic active site of aromatase, but also it makes a stable coordination with Fe atom of heme through an oxygen atom (O2). With such binding mechanism, this compound seems to be able to inhibit aromatase in the most effective way. Therefore, it may be proposed as the most potent inhibitor of aromatase and could be a new candidate for the treatment of breast cancer.
Acknowledgement
The author is grateful to School of Computer Science, Institute for Research in Fundamental Science (IPM), Tehran, Iran for professional technical assistance.
References
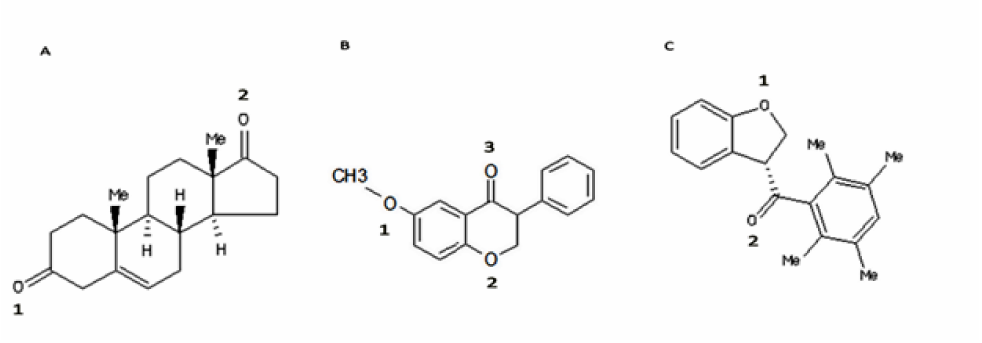
Figure 1: Chemical structure of androstenedione (ASD) as a natural substrate of Aromatase (A), 2a as an isoflavone derivative synthesized by Bonfield et al. (2012) with the most potent inhibitory effect (B), and 2a_1 with the lowest docking energy (-11.2 kcal/mol) against Aromatase among 286 compounds with 70 % of structural similarity to 2a which were chosen for structure-based virtual screening (C).
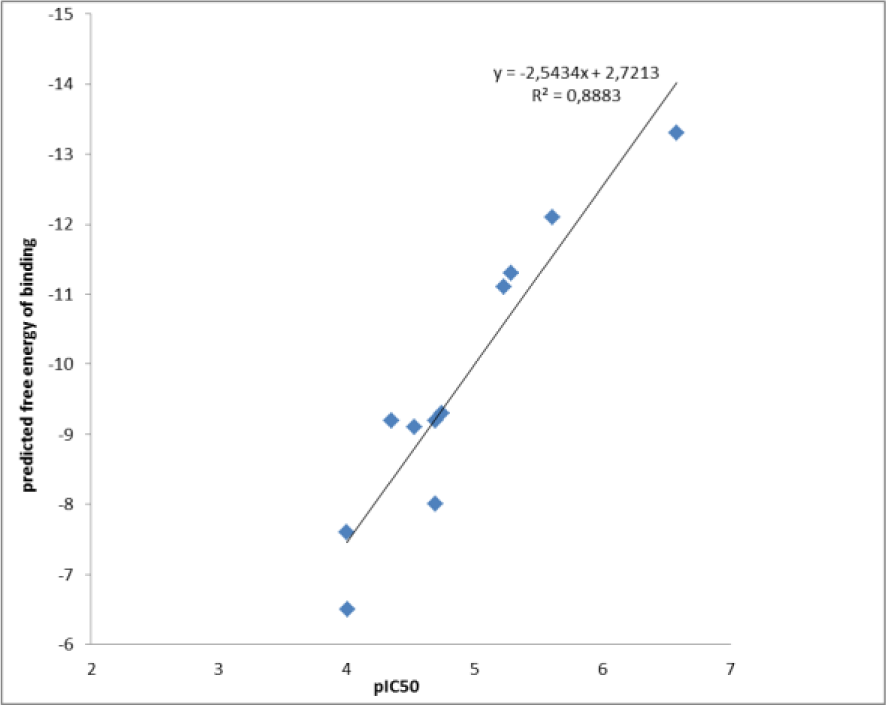
Figure 2: Correlation between estimated free energy of binding of aromatase inhibitors calculated by Autodock vina and experimental activities(log IC50). R2 for this correlation was 0.88.
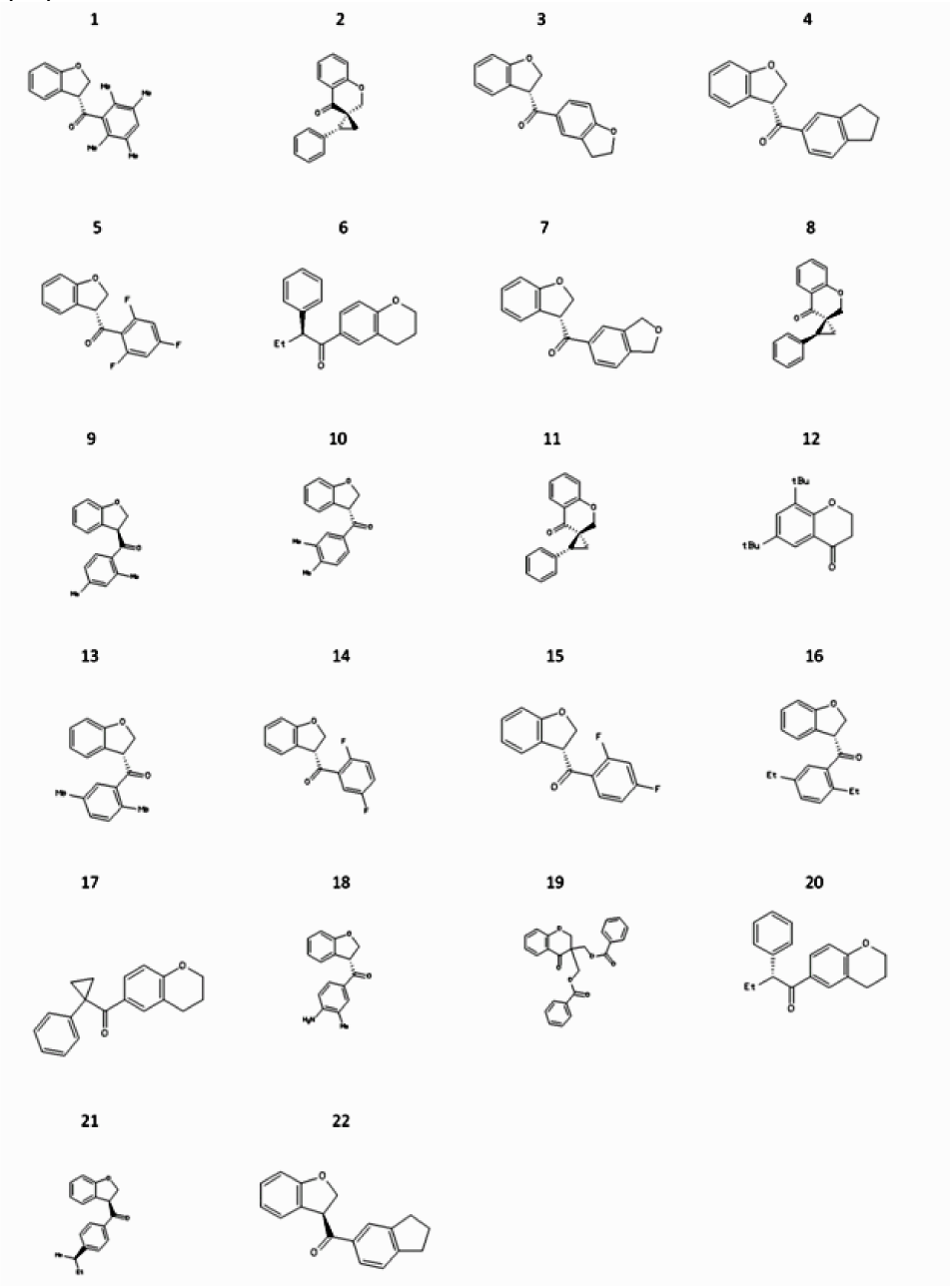
Figure 3: Among 286 chosen compounds from virtual screening which were docked against crystal structure of Aromatase, 22 of them had docking energies below -10 kcal/mol. The chemical structures of these compounds are depicted here. 2a_1 (Compound No. 1) by -11.2 kcal/mol has the lowest docking energy.
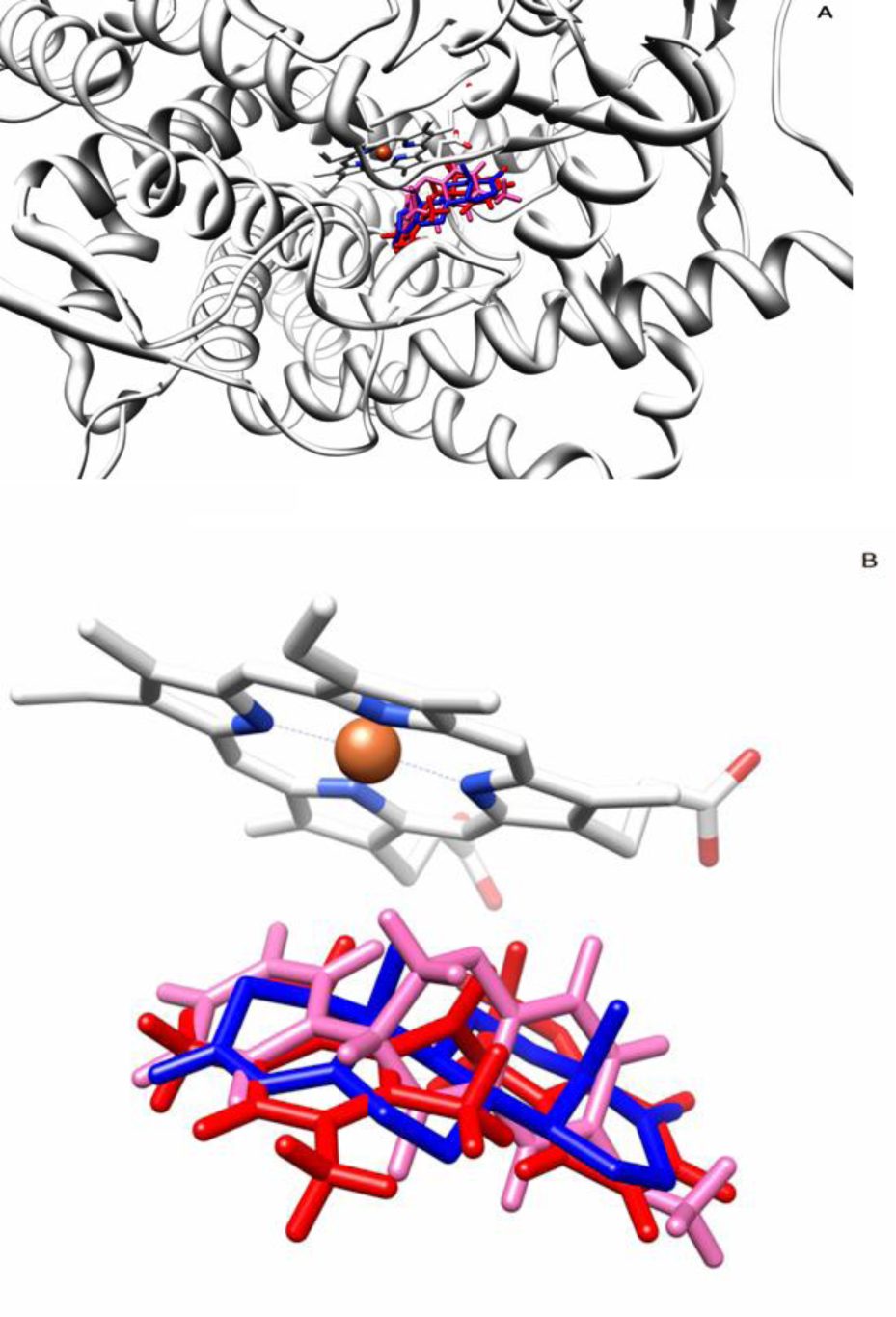
Figure 4: Superimposition of ASD (blue), 2a (pink) and 2a_1 (red) within the active site of Aromatase (A) and their position relative to the heme group of Aromatase (B).
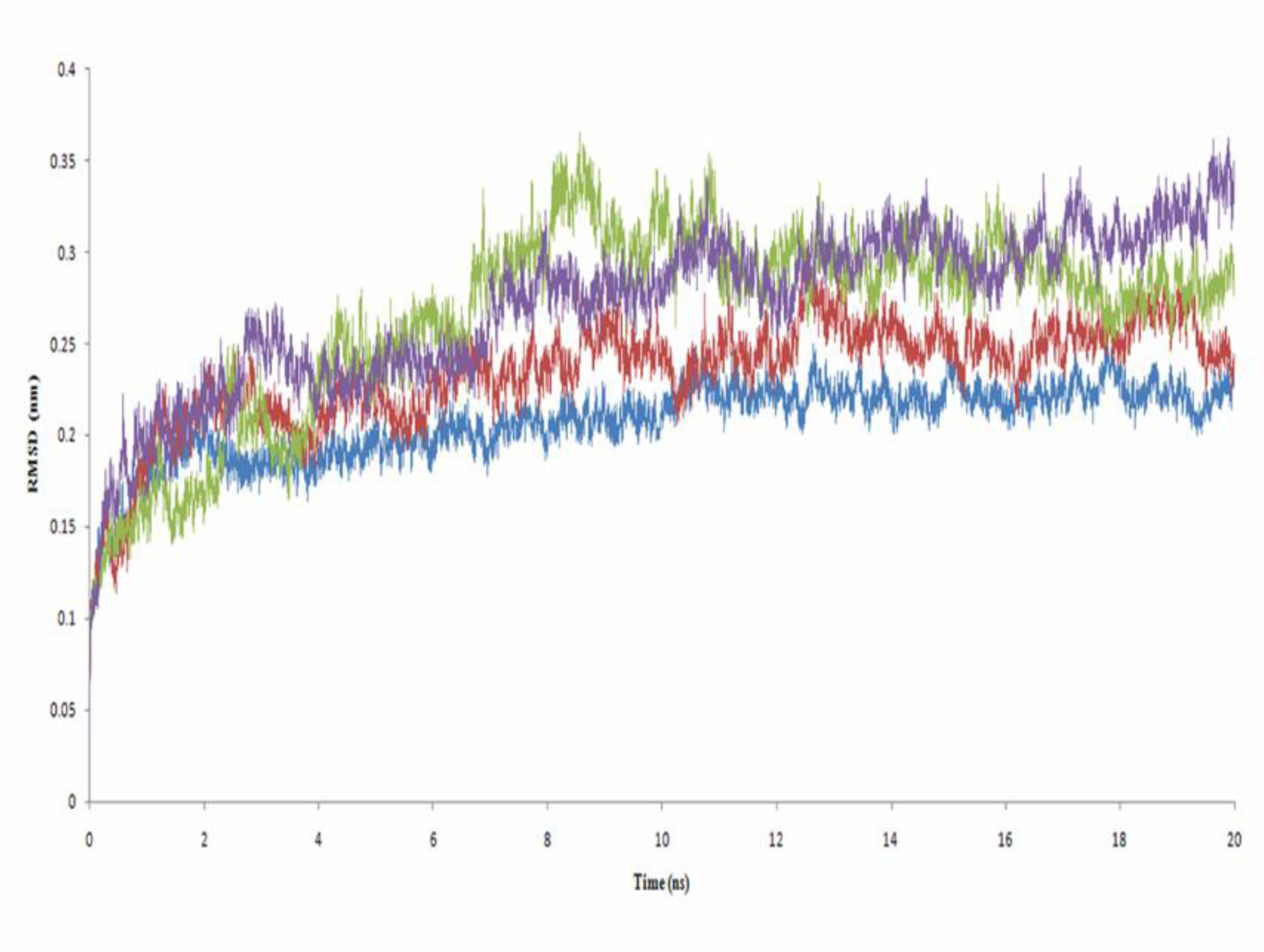
Figure 5: Root mean square deviation of Aromatase (blue line), Aromatase-ASD complex (red line), Aromatase-2a complex (green line) and Aromatase-2a_1 complex (purple line) during 20 ns of molecular dynamic simulation
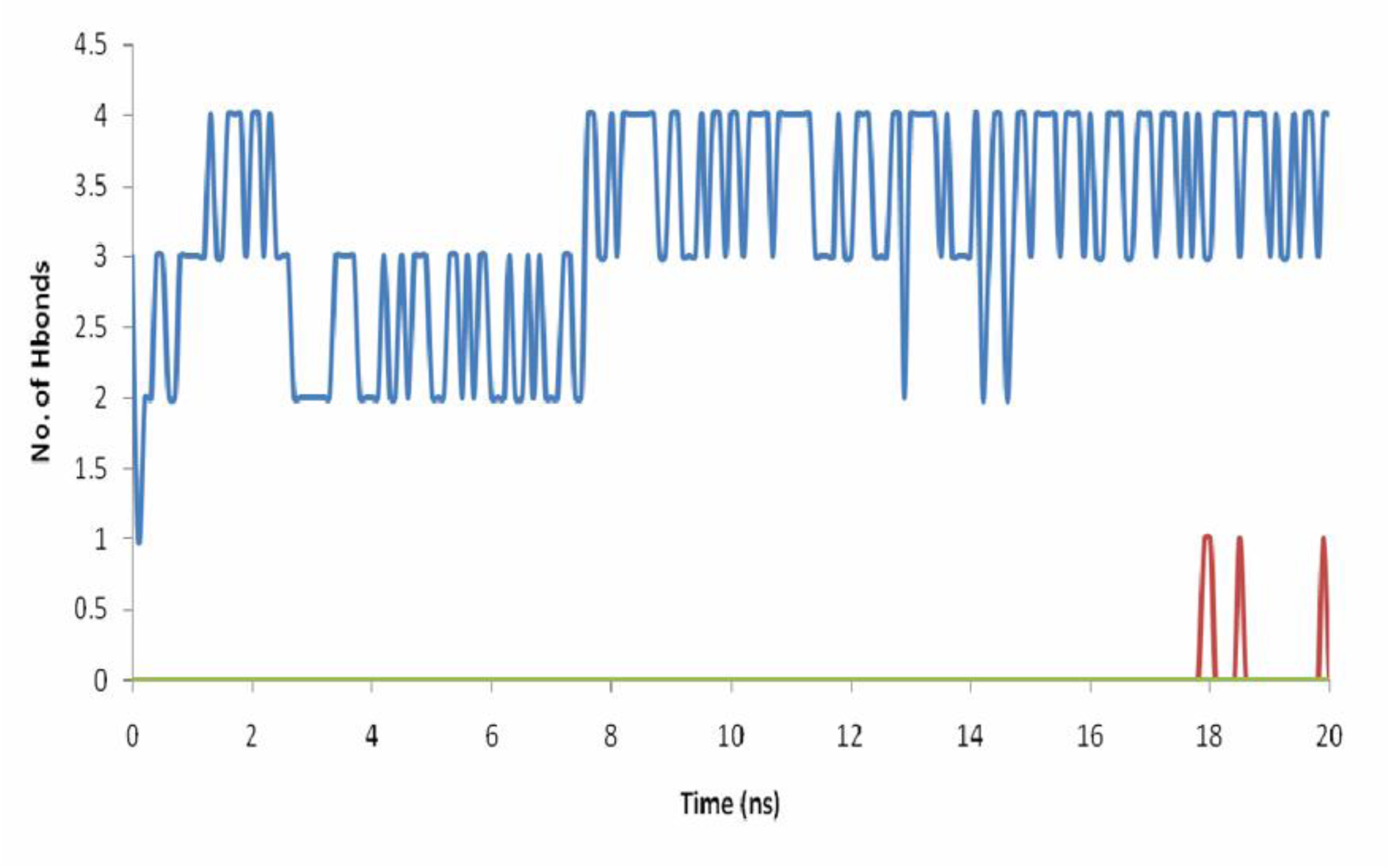
Figure 6: Number of hydrogen bonds between ASD (blue line), 2a (red line) and 2a_1 (green line) with Aromatase through 20 ns of MD simulation
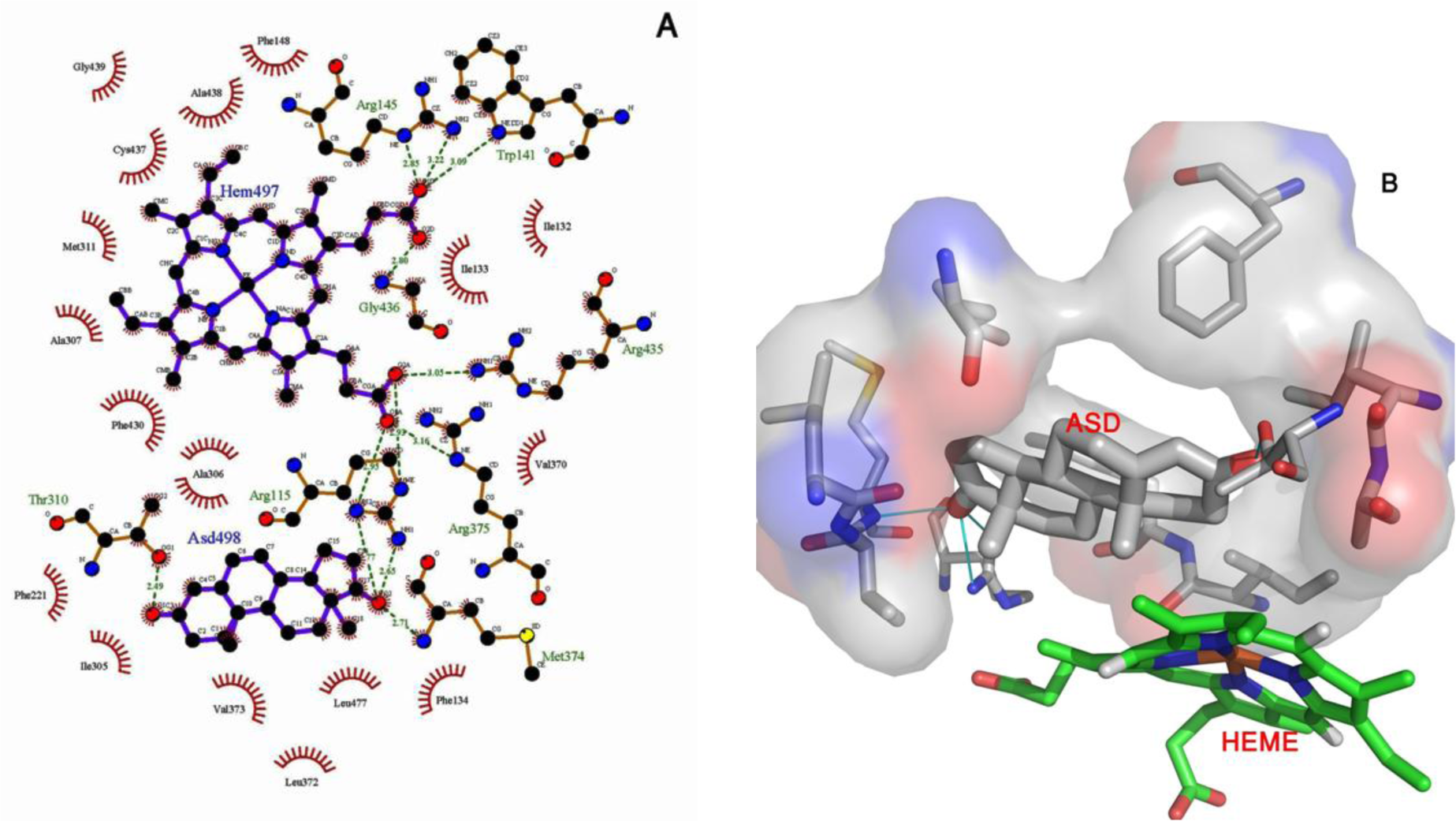
Figure 7: (A) 2D-image of ASD and heme interactions with Aromatase after 20 ns of MD simulation. As indicated, the hydrogen bonds formed between O1 from ASD with Thr310 and O2 of ASD with Met374 and Arg115 are the major interactions which result in the high affinity of ASD for Aromatase. The numbers indicate the minimum distance between heavy atoms that are involved in generated hydrogen bonds. (B) 3D-image of ASD with Aromatase residues which have a key role in clamping of ASD by Aromatase through hydrogen bonds and their position in relation to the heme group.
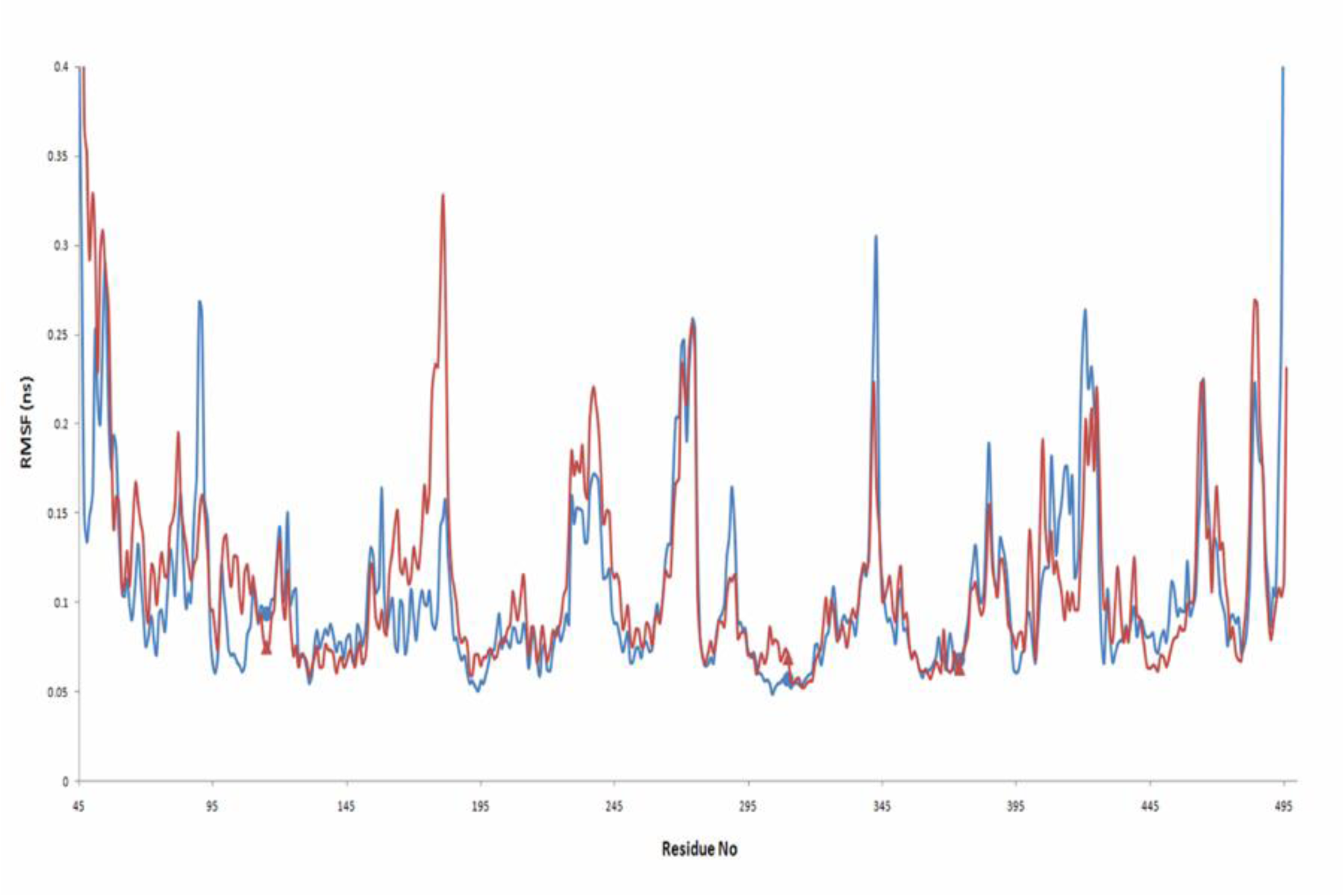
Figure 8: Root mean square fluctuation of alpha carbons from Aromatase residues as alone (blue line) and in complex with ASD (red line). RMSF of alpha carbons from Arg115, Thr310 and Met374 which interact with ASD via hydrogen bonds through 20 ns of MD simulation are indicated by the blue circle for Aromatase and by the red triangle for Aromatase-ASD complex.
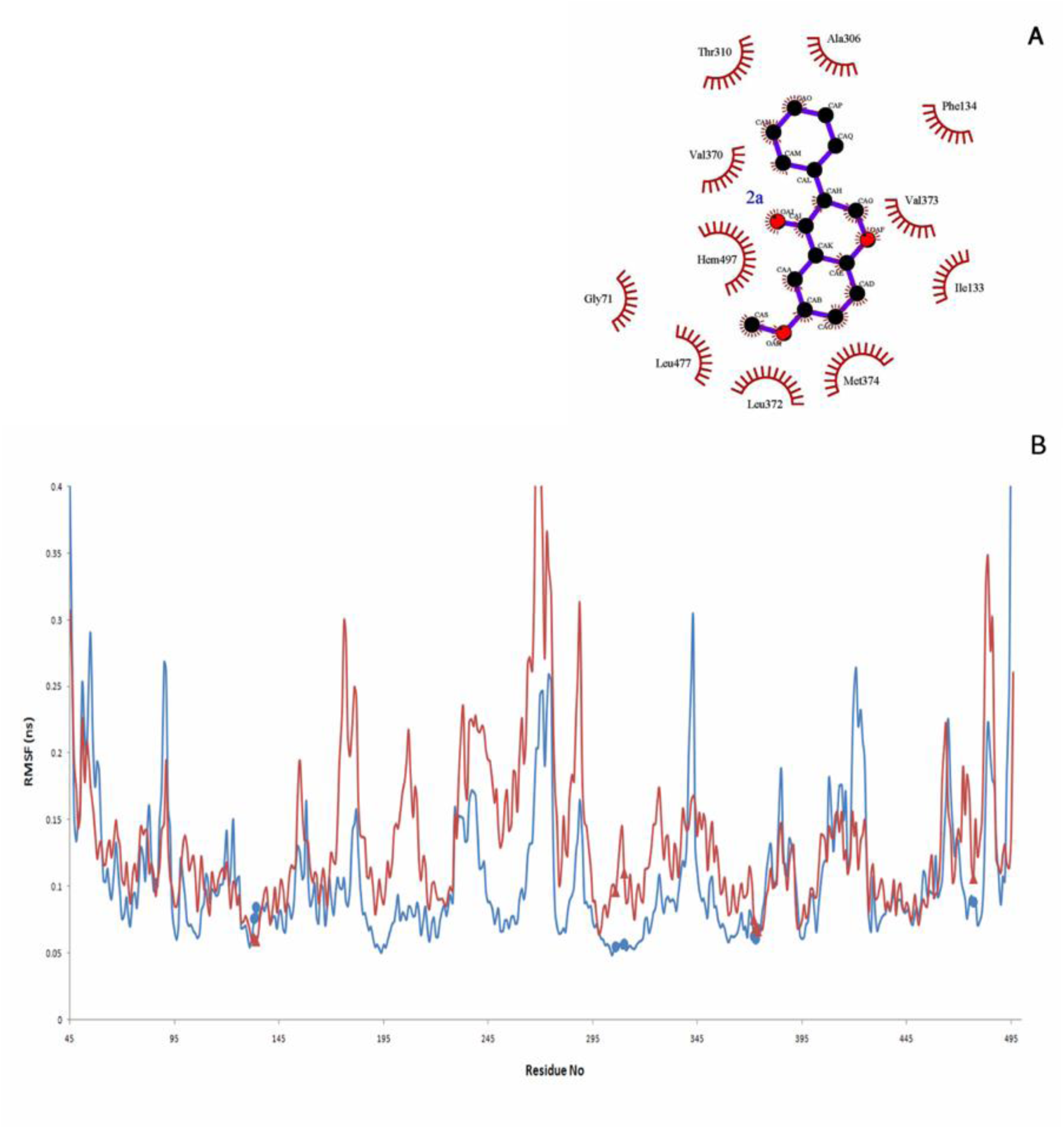
Figure 9: (A) 2D-image of Aromatase-2a interactions after 20 ns of MD simulation. 2a is surrounded by a hydrophobic tunnel, which is made by Ile133, Phe134, Ala306, Thr310, Leu372, Val 373, Met374 and Leu477. (B) The RMSF of alpha carbons of Aromatase (blue line) and Aromatase-2a residues (red line) through 20 ns MD simulation. Residues of Aromatase, which may interact with 2a through hydrophobic interactions are marked by blue circles. Same residues in Aromatase-2a complex are shown by red triangles.
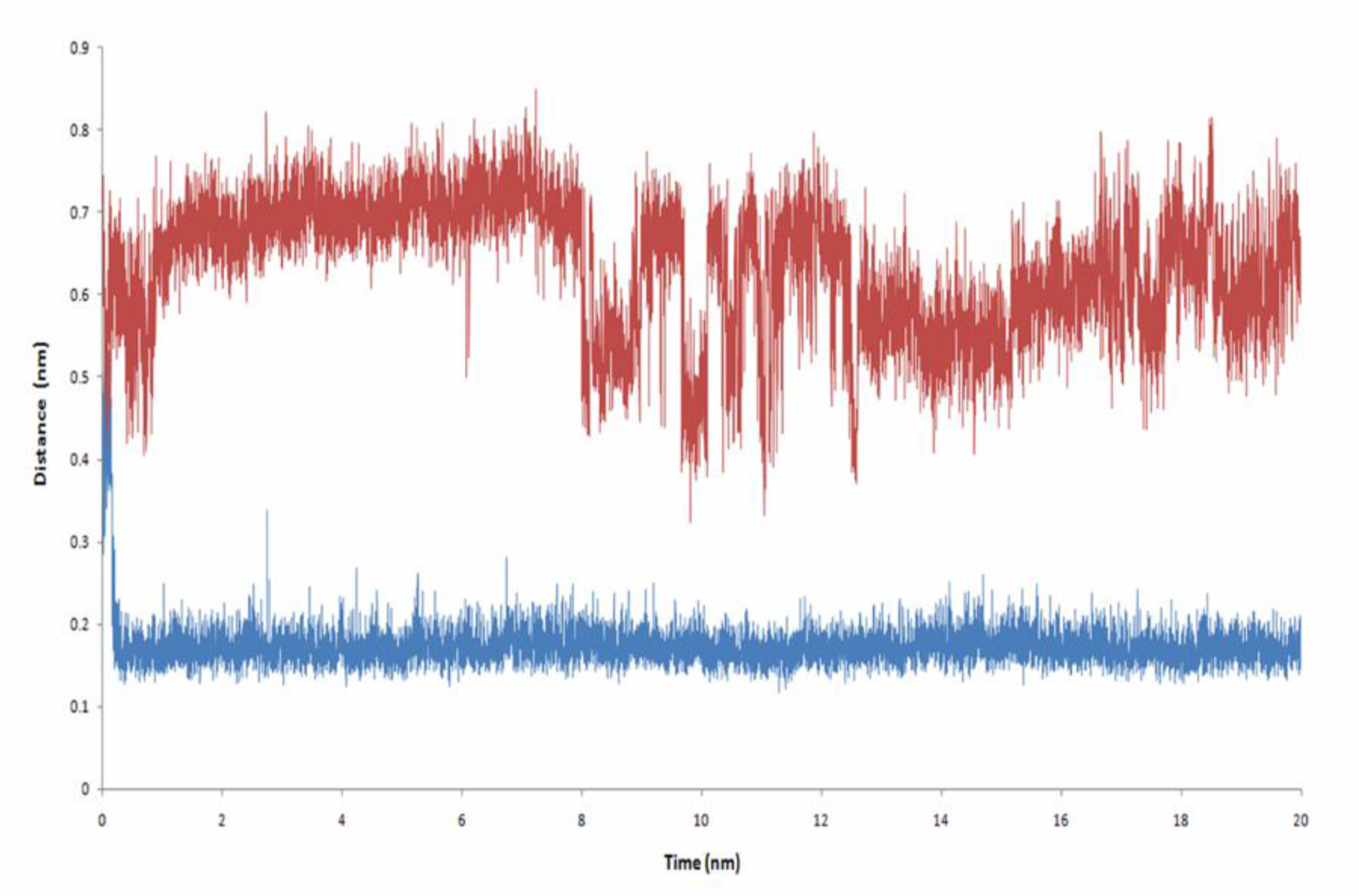
Figure 10: Distance between Iron atom from heme group of Aromatase with O2 of 2a (red line) and O2 of 2a_1 (blue line) through 20 ns of MD simulation. O2 from 2a_1 could coordinate in stable manner with Fe atom from the Aromatase, while O2 of 2a coordination with iron atom is so weak and unstable. Interaction of 2a_1 with aromatase via hydrophobic interactions and oxygen-iron coordination
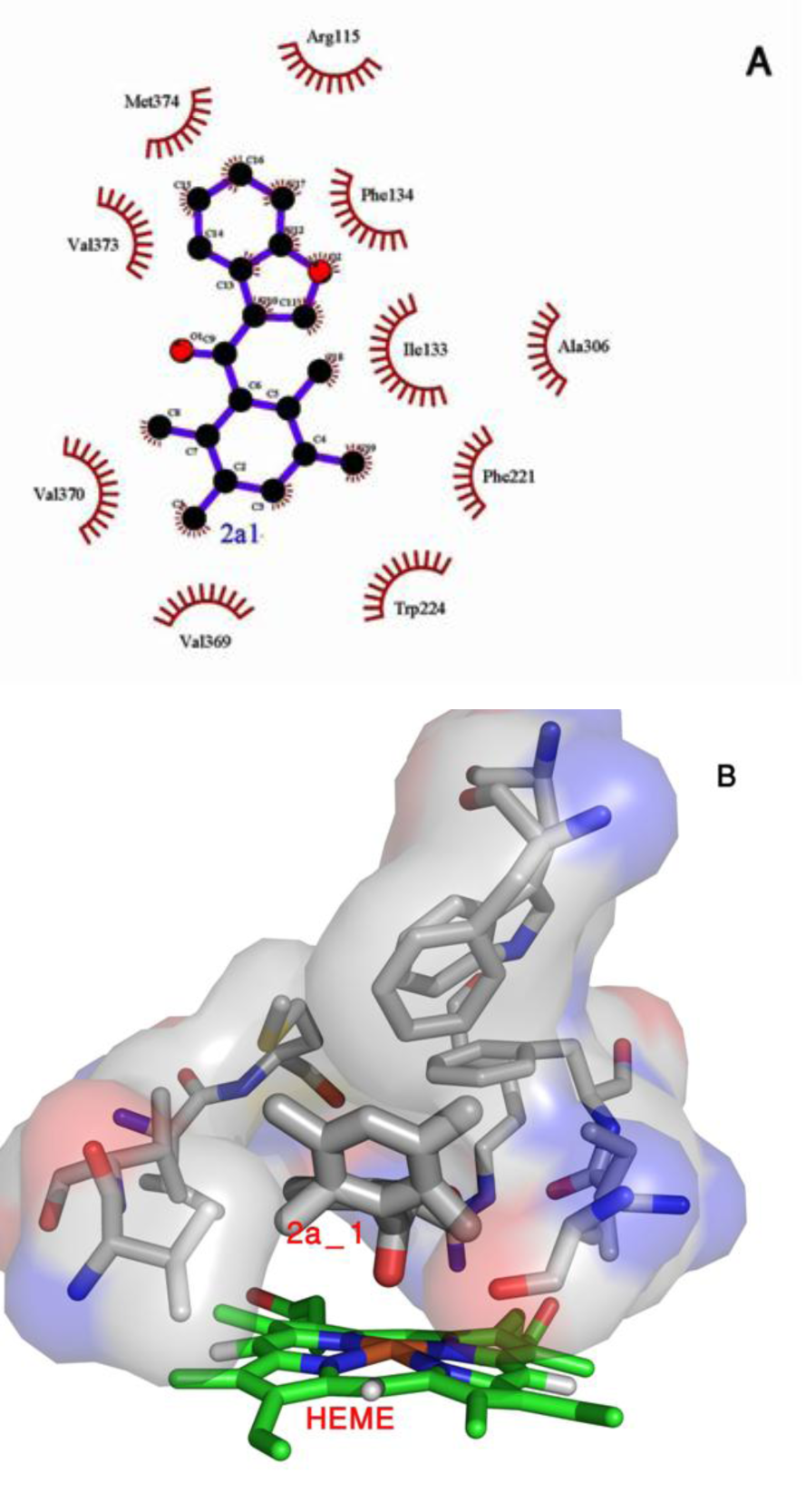
Figure 11: (A) 2a_1 after 20 ns of MD simulation surrounded by a hydrophobic hole which is made through Ile133, Phe134, Trp224, Ala306, Val373 and Met374 from Aromatase. (B) It seems that the high affinity of 2a_1 for Aromatase is mediated not only via hydrophobic interactions but also through a stable coordination, which forms between its O2 with Fe atom of heme group from Aromatase. This image shows the position of 2a_1 in relation to the heme group of Aromatase after 20 ns of simulation process.
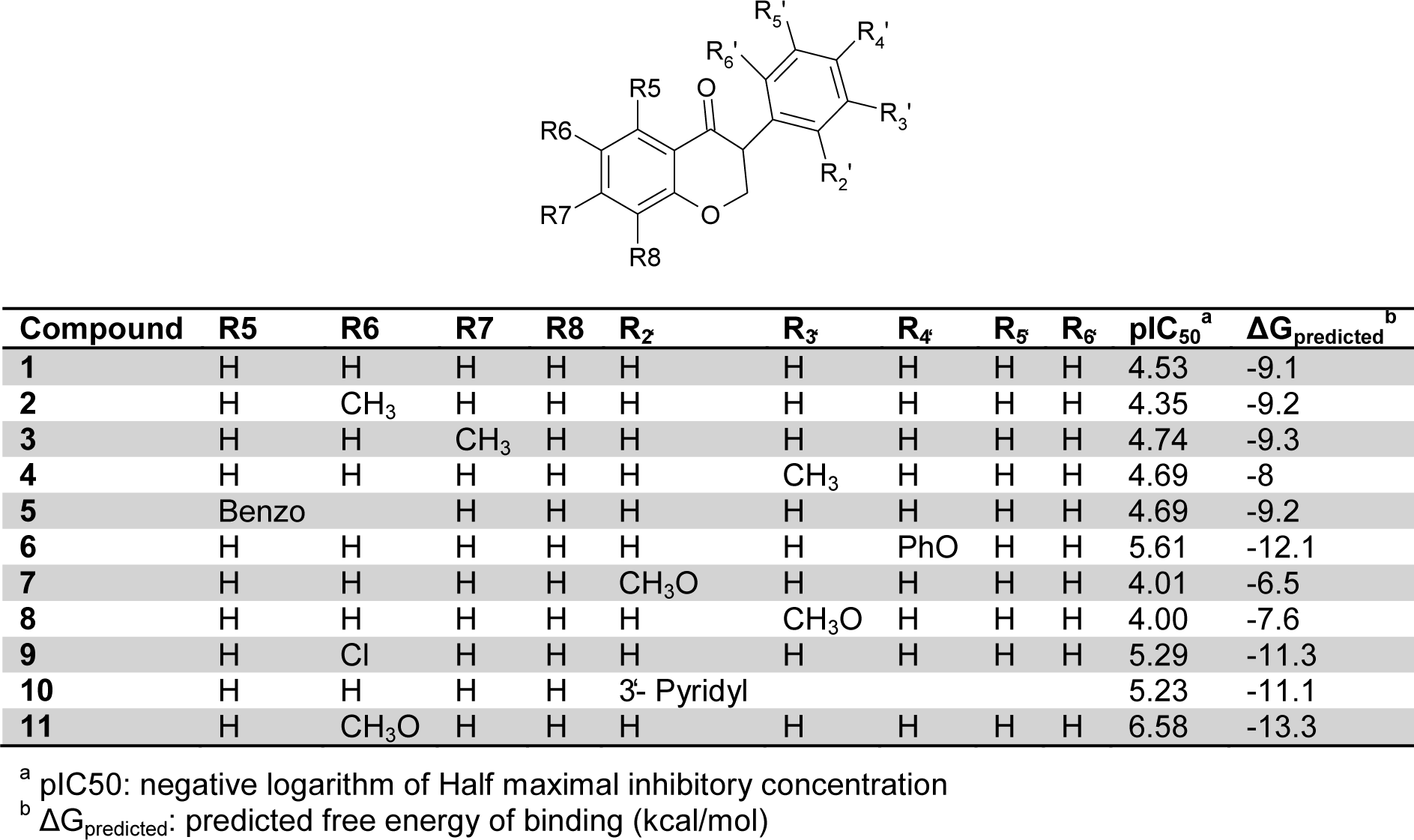
Table 1: Compound structures, free energy of docking and the negative logarithm of IC50 of eleven aromatase inhibitors. This structures were taken from literature (Bonfield et al., 2012)
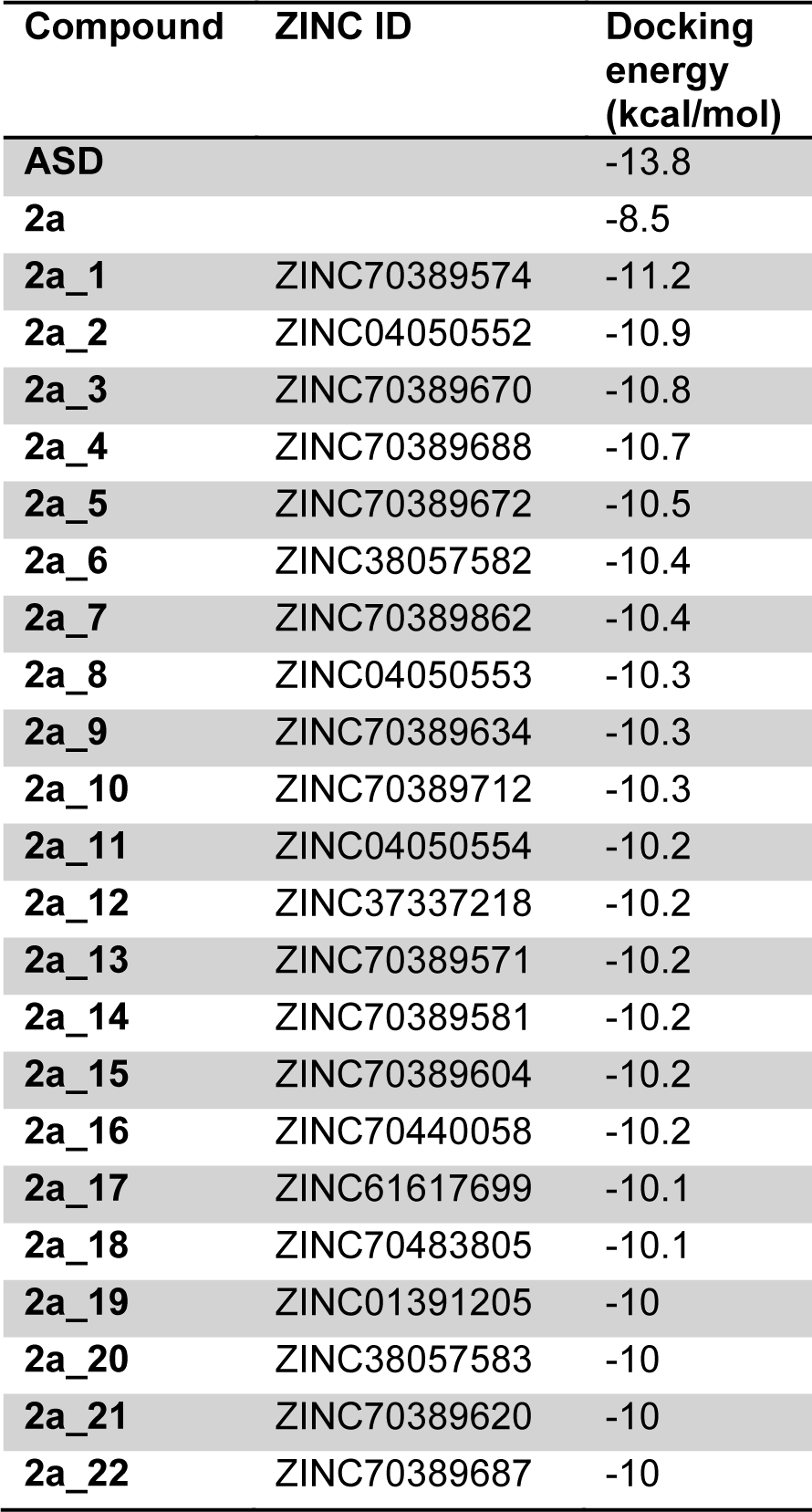
Table 2: Among all of 286 docked compounds with 70 % of similarity to 2a which were captured from ZINC server, just 22 of them had docking energy equal to or lower than -10 kcal/mol. These compounds are shown here.
[*] Corresponding Author:
Mostafa Jamalan, Science and Research Branch, Islamic Azad University, Ponak Square, Tehran, Iran. P.O. Box: 1477893855; Tel. No.: +98-611-5527199, Fax: +98-611-5527199, eMail: mjamalan@ibb.ut.ac.ir