Research article
Combined 3D-QSAR modeling and molecular docking study on multi-acting quinazoline derivatives as HER2 kinase inhibitors
Sako Mirzaie1[*], Majid Monajjemi2, Mohammad Saeed Hakhamaneshi3, Fardin Fathi4, Mostafa Jamalan1
1Department of Biology, Faculty of Science, Science and Research Branch, Islamic Azad University, Tehran, Iran2Department of Chemistry, Faculty of Science, Science and Research Branch, Islamic Azad University, Tehran, Iran
3Department of Biochemistry, Faculty of Medicine, Kurdistan University of Medical Science, Sanandaj, Iran
4Cellular and Molecular Research Center, Kurdistan University of Medical Sciences, Sanandaj, Iran
EXCLI J 2013;12:Doc130
Abstract
A series of new quinazoline derivatives has been recently reported as potent multi-acting histone deacetylase (HDAC), epidermal growth factor receptor (EGFR), and human epidermal growth factor receptor 2 (HER2) inhibitors. HER2 is one of the major targets for the treatment of breast cancer and other carcinomas. Three-dimensional structure-activity relationship (3D-QSAR) is a well-known technique, which is used to drug design and development. This technique is used for quantitatively predicting the interaction between a molecule and the active site of a specific target. For each 3D-QSAR study, a three-dimensional model is created from a large curve fit to find a fitting between computational descriptors and biological activity. This model could be used as a predictive tool in drug design. The best model has the highest correlation between theoretical and experimental data. Self-Organizing Molecular Field Analysis (SOMFA), a grid-based and alignment-dependent 3D-QSAR method, is employed to study the correlation between the molecular properties and HER2 inhibitory potency of the quinazoline derivatives. Before presentation of inhibitor structures to SOMFA study, conformation of inhibitors was determined by AutoDock4, HyperChem and AutoDock Vina, separately. Overall, six independent models were produced and evaluated by the statistical partial least square (PLS) analysis. Among the several generated 3D-QSARs, the best model was selected on the basis of its statistical significance and predictive potential. The model derived from the superposition of docked conformation with AutoDock Vina with reasonable cross-validated q2 (0.767), non cross-validated r2 (0.815) and F-test (97.22) values showed a desirable predictive capability. Analysis of SOMFA model could provide some useful information in the design of novel HER2 kinase inhibitors with better spectrum of activity.
Keywords: quinazoline, human epidermal growth factor receptor 2, 3D-QSAR, self-organizing molecular field analysis, AutoDock
Introduction
The epidermal growth factor receptor (EGFR) is the cell-surface receptor of the epidermal growth factor family (Herbst, 2004[17]). This family comprises four structurally related tyrosine kinase receptors, namely, epidermal growth factor receptor 1 (alternatively named EGFR, ErbB1) or human epidermal growth factor receptor 1 (HER1), HER2 (ErbB2), HER3 (ErbB3) and HER4 (ErbB4) (Cho et al., 2003[10]; Satyanarayanajois et al., 2009[27]; Woodburn, 1999[33]; Yarden et al., 2004[34]). ErbB receptors are made up of an extracellular region or ectodomain that provides a ligand-binding site, a single transmembrane-spanning region, and a cytoplasmic tyrosine kinase domain (Garrett et al., 2002[14]). When a certain ligand binds to the extracellular domain of an ErbB receptor, its dimerization is triggered with other adjacent ErbB. There is not any known ligand for HER2 and instead this receptor acts as a co-receptor for each of the other three (Linggi and Carpenter, 2006[21]). Dimerization leads to a rapid activation of receptor's cytoplasmic kinase domain (Satyanarayanajois et al., 2009[27]). Activated form of the ErbB receptors plays a critical role in the regulation of cell growth, proliferation and migration. Kinase domain subsequently catalyzes receptor autophosphorylation and tyrosine phosphorylation of ErbB substrates (Satyanarayanajois et al., 2009[27]; Pawson, 1995[24]). It has been shown that HER2 overexpression is involved in colon, ovarian, non-small-lung cell and approximately 25-30 % of breast cancers (Shmeeda et al, 2009[29]). Furthermore, amplification content is conversely correlated with the observed average survival time (Carter et al., 1992[8]).
Breast cancer is one of the major health problems among developed community. It is the most common neoplasia among women, in which more than 1 million new cases occur every year, and it is the first cause of death in women aged 40-59 years old (Galvao et al., 2011[13]). Just in 2010, 207,090 new cases are estimated to occur in United States with an expected mortality rate of 39,840 women (Galvao et al., 2011[13]; Jemal et al., 2010[18]). Obstruction of HER2-mediated multimerization leads to inhibition of phosphorylation and subsequently, uncontrolled cell growth and division will limit. So, blocking of HER2-mediated signaling has made it a key target of breast cancer therapies (Satyanarayanajois et al., 2009[27]). Trastuzumab (Herceptin) is an FDA-approved humanized monoclonal antibody, which is used to treat the early stage of HER2 overexpressed breast cancer. It binds to the ectoplasmic domain of HER2 and inhibits any receptor dimerization (Burgess et al., 2003[6]). Use of trastuzumab as an effective drug, because of its cost, is still controversial. In the U.S., 34-week treatments for every case (average duration of treatment) costs approximately 23,400 USD (Stebbing et al., 2000[30]). Recent studies have also shown that cancer treatment by trastuzumab may be contributed in one kind of drug-induced cardiac dysfunction (Zeglinksi et al., 2011[35]). In contrast with trastuzumab, chemical new inhibitors like erlotinib and lapatinib were designed and synthesized against cytoplasmic kinase domain of HER2 (Cai et al., 2010[7]). Because of high identity and similarity between HER2 kinase domain and the other more than one hundred kinases in cells, a highly specific kinase domain inhibitor is challenging. On the other side, drug resistance could occur due to ATP-binding site mutations (Satyanarayanajois et al., 2009[27]). So, it seems that the need to design and synthesis of a more potent and specific HER2 kinase inhibitor is urgent.
Quantitative structure-activity relationships (QSAR) have played an influential role in the design of pharmaceuticals in medicinal chemistry (Akamatsu, 2002[3]). In this technique, variation in biological activities is statistically analyzed using descriptors related to structural properties of compounds. The self-organizing molecular field analysis (SOMFA) is a simple three-dimensional quantitative structure-activity relationship (3D-QSAR) technique. In this method, the molecular shape and electrostatic potential are used to construct the QSAR models (Robinson et al., 1999[26]). Recent study reports the first published crystal structure of the kinase domain of HER2, which could be a valuable target to design the new anti-HER2 drugs (Aertgeerts et al., 2011[1]). The purpose of this paper is to describe the application of molecular docking studies and self-organizing molecular field analysis, SOMFA, on a series of quinazoline derivatives. The worthwhile molecular architecture required for designing new specific inhibitors against HER2 positive breast cancer is also discussed.
Materials and Methods
Overall strategy
To produce the SOMFA model, minimized and correct geometrical conformations of quinazoline derivatives are needed. For this purpose, three independent categories of inhibitor conformations were produced by AutoDock 4.2 (Morris et al., 2009[23]), HyperChem 8.0 (Froimowitz, 1993[12]) and AutoDock Vina (Trott and Olson, 2010[32]). For each category, compound 1 was selected as the reference compound and the other compounds were superimposed by atom based alignment technique (Figure 1(Fig. 1)). In this method, superposition of molecules is based on trying to minimize root-mean-squares (RMS) differences in the fitting of selected atoms with those of a reference molecule (Figure 2(Fig. 2)). Finally, for each category, related SOMFA models were produced by SOMFA software (Figure 3(Fig. 3)) (Robinson et al., 1999[26]).
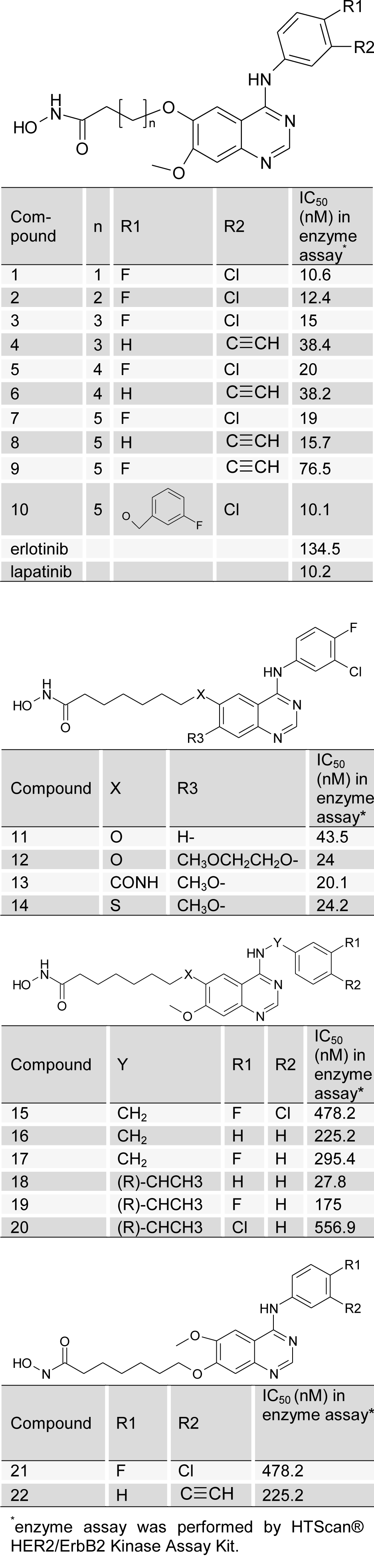
In this study, a data set of 24 compounds belonging to the quinazoline derivatives as HER2 inhibitors were taken from the literature (Cai et al., 2010[7]) and used to produce SOMFA model (Table 1(Tab. 1), Cai et al., 2010[7]). The chemical structures of mentioned compounds were generated by PRODRG server (Schuttelkopf and van Aalten, 2004[28]). For optimization of constructed compounds, molecular mechanic (MM+) optimization followed by the semi-empirical AM1 (Austin Model 1) method implemented in HyperChem software were used. For both the above mentioned methods, molecular structures were optimized using the Polak-Ribiere (conjugate gradient) algorithm until the root mean square (RMS) gradient achieves a value smaller than 0.001 kcal mol-1.
Biological activity
The negative logarithm of the measured IC50 (M) against human HER2 kinase domain as pIC50 was used as dependent variable (Cai et al., 2010[7]). HER2 kinase activity was measured using HTScan® HER2/ ErbB2 Kinase Assay Kit (Cell Signaling Technology). It includes active HER2/ErbB2 kinase (supplied as a GST fusion protein), a biotinylated peptide substrate and a phospho-tyrosine antibody for detection of the phosphorylated form of the substrate peptide. The enzyme assay was performed based on fluorescent immuno-detection approach (Cai et al., 2010[7]).
Protein preparation
3D crystallographic structure of the kinase domain of human HER2 (ErbB2) with PDB code 3PPO (Aertgeerts et al., 2011[1]) was obtained from the Brookhaven protein data bank (Berman et al., 2000[4]). For use of kinase domain structure in docking simulation, all missing hydrogen atoms were added by Hbuild command implemented in CHARMM molecular dynamic package (Brooks et al., 2009[5]). All water molecules were eliminated except for water ID 22, which is tightly bounded to the active site of HER2 kinase domain. After initial minimization by Adopted Basis Newton-Raphson (ABNR) and steepest descent (SD) methods, output structure was saved and used for molecular docking studies.
Molecular docking studies
AutoDock 4.2
Autodock is a flexible docking technique which is based on Monte Carlo simulated annealing search to find the optimal conformation of ligand in a macromolecule. Inhibitor structures minimized by HyperChem were used as input ligand files for AutoDock 4.2. Ligand preparation was done by AutoDockTools. The grid maps were calculated using AutoGrid (part of the AutoDock package). Because the active site of HER2 kinase domain is known (Aertgeerts et al., 2011[1]), a grid map with 25 × 25 × 25 points with a grid-point spacing of 1 Å (roughly a quarter of the length of a carbon-carbon single bond) were defined. For both protein and ligands, the partial atomic charges were calculated using the Gasteiger-Marsili method (Gasteiger and Marsili, 1980[15]). For all dockings, 50 independent runs with the step sizes of 0.2 Å for translations and 5° for orientations and torsions were performed. For each ligand, pose with the minimum free energy of binding was saved and used for SOMFA study.
AutoDock Vina
AutoDock Vina is a molecular docking software that profits of knowledge-based potentials and empirical scoring functions (Trott and Olson, 2010[32]). Minimized inhibitor structures were defined as input files for AutoDock Vina. The docking site for inhibitors on 3PPO was defined by setting up a box with a grid map with 25 × 25 × 25 points (grid-point spacing of 1 Å) at the geometrical center of the native ligand present in PDB structure. Software parameter "Exhaustiveness", which determines how comprehensively the program searches for the lowest energy conformation, was set to the default value, eight for all docking runs. For each docking calculation, structure with relative binding energy, based on the software cluster, was saved and employed to SOMFA study.
SOMFA 3D-QSAR study
To produce a 3D-QSAR model, SOMFA2 package software was used. This suite of programs is designed to enable the user to carry out an advanced 3D-QSAR analysis of a series of molecules. The results of the analysis enable the rationalization of the activities of known molecules together with the potential for reliably estimating the activity of molecules that have yet to be synthesized (Robinson et al., 1999[26]). In our SOMFA study, a 40 × 40 × 40 Å grid originating at (-20, -20, -20) with a two resolution (1 and 0.5 Å) were generated around the aligned compounds.
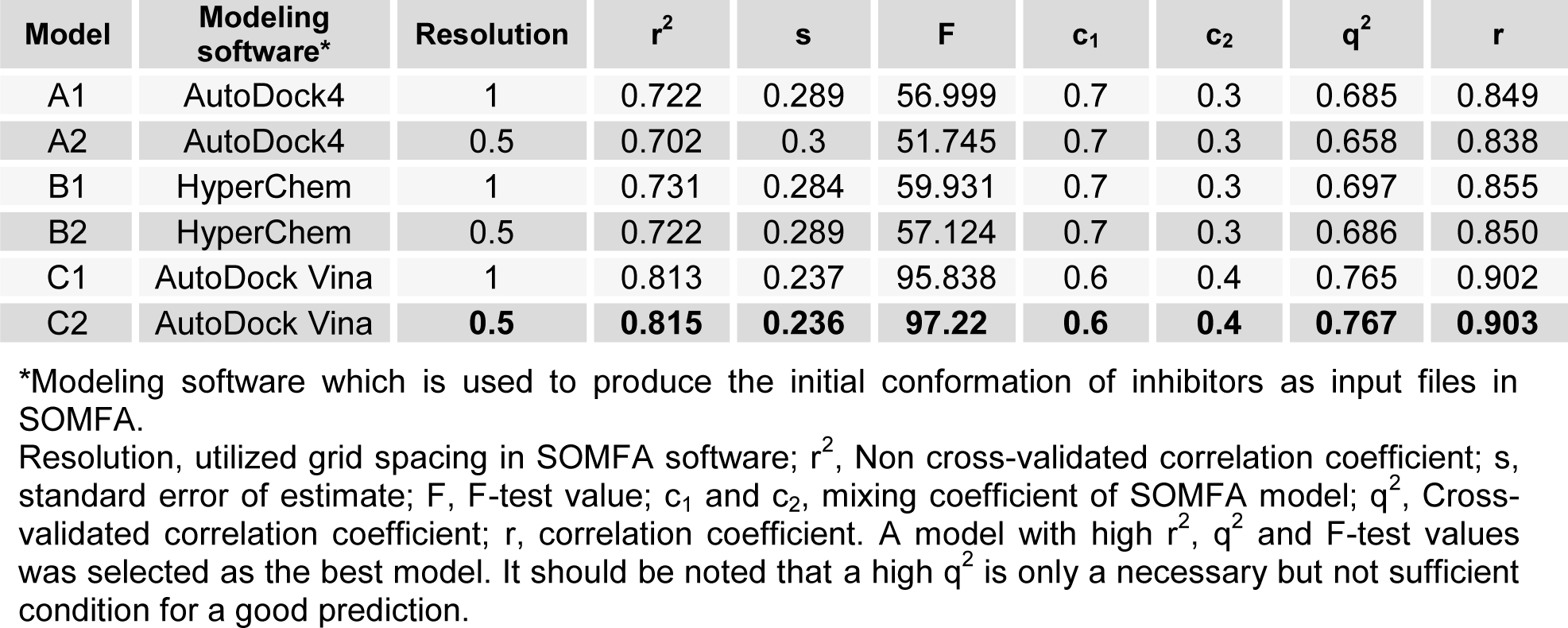
Six different models using different resolutions and aligned conformations of inhibitors have been produced and presented in Table 2(Tab. 2). For all the studies, shape and electrostatic potential were produced as an independent variable. These two variables, consequently, combined to create the final model by utilizing the partial least square (PLS) algorithm in conjugation with leave one out (LOO) cross-validation method. Based on equation 1, weighted average of shape and electrostatic were calculated:
Activity = c1 Activityshape + (1 - c1) ActivityESP (1)
where c1 is a mixing coefficient.
The predictive ability of the model is quantitated in terms of q2 (r2cv) which is defined in equation 2:
q2 = 1 - PRESS/SSD (2)
where SSD is the sum of squared deviations between observed values and the mean of predicted property values. PRESS is the sum of squared deviations between observed and predicted property values:
PRESS = ∑ (Yobserved - Ypredicted)
q2 takes up values in the range from 1 to less than 0. Low or negative values of q2 indicate the errors of prediction are greater than the error from assigning each compound mean activity of the model. Higher values of q2 is necessary but not sufficient for evaluating a QSAR model (Golbraikh and Tropsha, 2002[16]). One of the several variance-related parameters that can be used as a criterion of the level of statistical significance of the regression model is Fischer statistic parameter (F-value). The larger value of F implies that a more significant correlation has been reached (Kulkarni et al., 1999[19]). Because the final equations are not very useful to efficiently denote the SOMFA models, 3D master grid maps of the best models are displayed by Grid-Visualizer program. These grids represent an area in space where steric and electrostatic field interactions are responsible for the observed variations of the biological activity.
Results and Discussion
Numerous small organic compounds have been synthesized and evaluated as inhibitors of EGFR and HER2 tyrosine kinases (Traxler et al., 1997[31]; de Bono and Rowinsky, 2002[11]). Since, quinazoline derivatives could inhibit the HER2 and account for potent HER2/EGFR inhibitors (Cai et al., 2010[7]), 3D quantitative structure activity relationship study with aid of molecular docking method was done to develop more influential HER2/EGFR mediated anti-cancer drugs. In the first phase of this study, to ensure the docking protocol is set up correctly; an internal validation phase was done.
Internal validation phases
In this phase, SYR127063, which is the reference ligand of the crystal structure with PDB code 3PPO, was docked by AutoDock 4 and AutoDock Vina, separately (Aertgeerts et al., 2011[1]). Superimposing the experimental and predicted conformations was expressed as root mean square deviation (RMSD). Calculated RMSD for SYR127063 were 3.56 and 0.65 Å for AutoDock4 and AutoDock Vina, respectively. The results showed that the molecular docking method by AutoDock Vina is robust and suitable for estimating the interactions of such ligand with kinase domain of HER2. Conversely, computed RMSD of 3.56 is accepted as poor docking method. Figure 4(Fig. 4) shows the similarity between experimental and docked structures by two docking methods.
Models produced by 3D-QSAR
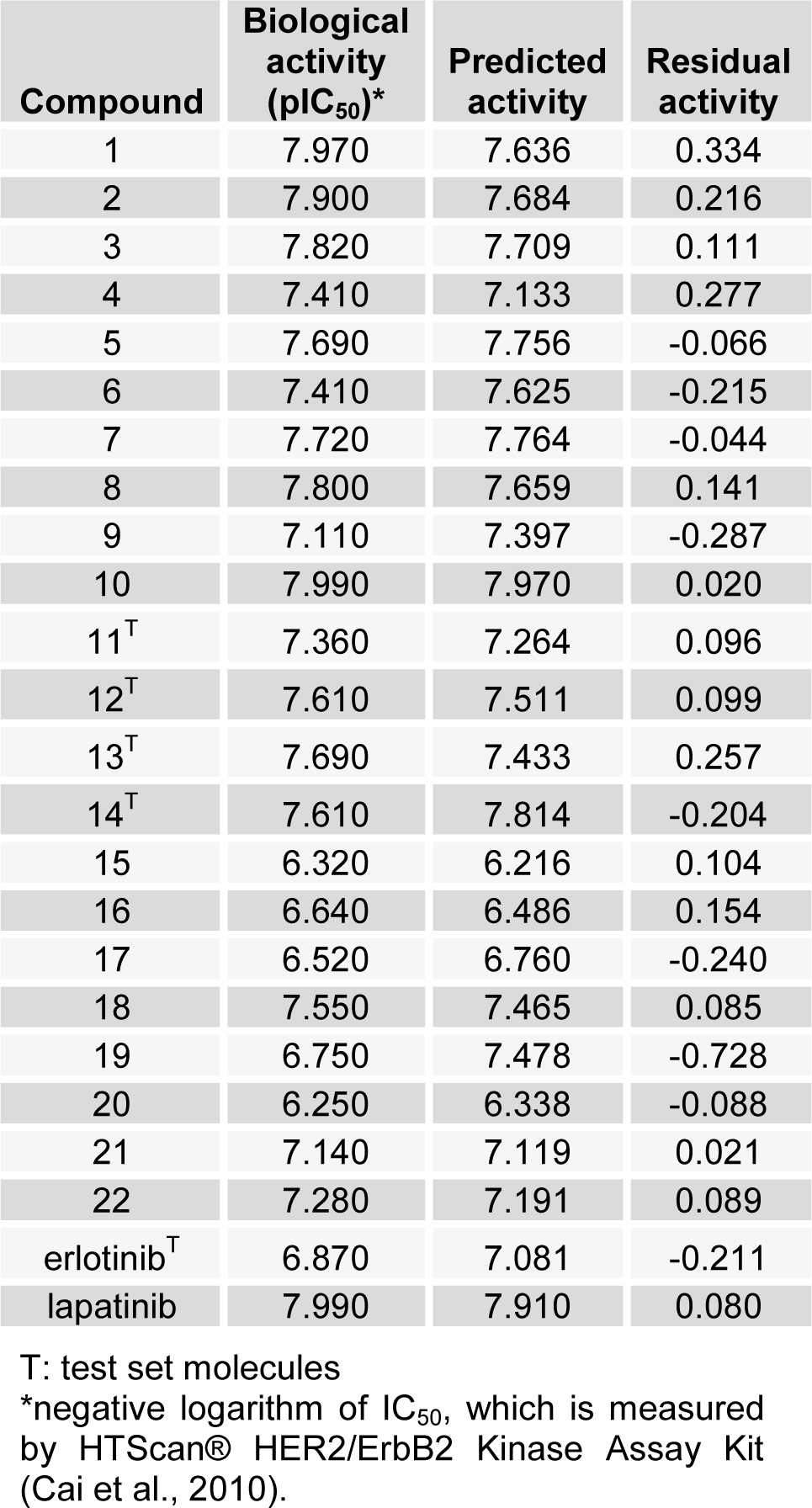
In this section, as mentioned prior, six models were produced based on initial coordination of inhibitors. For predicted models, cross-validated r2 or q2 and also F-values were computed and served as a quantitative measure of the predictability of the SOMFA models. Statistical analyses of models are summarized in Table 2(Tab. 2). Poor models are belonging to model A1 and A2 with the minimum values of q2 and high quantity of standard error. As shown in Figure 4(Fig. 4), the RMSD between docked and crystal structure is high, and it's not acceptable as a successful docking in molecular modeling studies. So, it can be postulated that the conformation of models, which are calculated by AutoDock4 have lower validity. When the three-dimensional structure of macromolecule target is not determined, or if it has not an acceptable resolution, or also when a critical amino acid of target active site is missed, use of semi-empirical methods to calculate the initial coordination in the 3D-QSAR study is common. Some studies apply this method instead of molecular docking (Rajwade and Pande, 2008[25]; Aggarwal et al., 2010[2]). Model B1 and B2 have higher values of q2. The minimization of structures of these models was done by HyperChem. Among all of models, the best one which is predicted by using of AutoDock Vina and SOMFA software was C2. The mentioned model which has q2 value of 0.76, r2 value of 0.8, minimum standard error of 0.23 and F-test value of 97.22, demonstrates a satisfying statistical correlation and predictive ability. Based on our results, the models with the initial structures docked by AutoDock Vina are the best and those docked with AutoDock4 are poor and models B1 and B2 are moderate in view of validity. In molecular docking software, three parameters determine the success of docking study. The first is ligand and macromolecule representation, which is same in both AutoDock4 and Vina. Second is the scoring function of docking software. AutoDock4 uses five terms, including van der Waals, electrostatic, hydrogen bond, torsional penalty and desolvation parameter as its scoring function (Morris et al., 2009[23]; Chang et al., 2010[9]). Instead, AutoDock Vina employs just three score function terms comprising hydrophobic interaction (van der Waals), hydrogen bond and torsional penalty (Trott and Olson, 2010[32]). Although, the above-mentioned docking packages use the same empirically-weighted scoring functions, but they have different empirical parameters and thereby, have a distinctive calculation. The third item that affects docking validity is the search algorithm. With regard to Figure 4(Fig. 4) and Table 2(Tab. 2), it seems that stochastic search algorithm of AutoDock4 has a poor function and efficiency compared to Vina. AutoDock Vina optimizes its local search application by calculation of derivatives of any ligand conformation energy, which is known as the gradient-based local search algorithm. Such method could increase speed and accuracy in AutoDock Vina. Trott and Olson (2010[32]) showed that in re-docking of 190 protein-ligand complexes with RMSD tolerance of 2 Å, success percentage of AutoDock4 and Vina was 49 % and 78 %, respectively (Trott and Olson, 2010[32]). Statistical analysis of our data and also the calculated RMSD of SYR127063 in validation phase confirm such conclusion. In the course of SOMFA study, grid spacings of 1 and 0.5 Å were examined. Our data showed that in comparison with 1 Å, the 0.5 Å grid spacing generates a model with a good correlation (Table 2(Tab. 2)). For the other models, it seems that 1 Å grid spacing is suitable. It can be postulated if the structures with wrong or far from native conformation are used for grid-based 3D-QSAR, with decreasing of grid spacings, the amount of noise in the descriptor data is increased. Therefore, the quality of the model is reduced. In model C2, which has the best correlation with experimental data, the biological and predicted activities of the training and test sets are presented in Table 3(Tab. 3). Figure 5(Fig. 5) is depicted based on Table 3(Tab. 3) and shows a good correlation (r2= 0.81) between biological and predicted activity of HER2 inhibitors. Figure 6(Fig. 6) shows the plot of the predicted versus biological activity values of test set, demonstrating that the most compounds are predicted satisfactorily.
Electrostatic and shape contribution in SOMFA model
SOMFA produces a model based on shape and electrostatic independent variables. Shape values are given a value of 1 inside the van der Waals envelope and 0 for outside. Electrostatic values calculated by use of the partial charges distributed across the atom centers (Robinson et al., 1999[26]) the SOMFA models. The models that have been produced using AutoDock 4 or Vina, electrostatic contribution was decreased to 0, by use of Gasteiger charge (data not shown). However, by assigning the AM1 charge as a partial atom charge, this proportion was raised. The same deduction was presented by Li et al. (2011[20]). It has been shown that the partial charge computation method can affect the validity of 3D-QSAR models in a statistically significant mode (Mittal et al., 2009[22]). AM1 charges are a set of Mulliken-type charges derived from a semi-empirical quantum-mechanical calculation (Mittal et al., 2009[22]). Semi-empirical quantum methods are a mediocre between the empirical and ab initio quantum chemical approaches in terms of accuracy and computational time. Instead, Gasteiger charge calculation is an empirical atomic partial charge method which utilizes experimental parameters of its data set (Gasteiger and Marsili, 1980[15]). One of the reasons for superiority of AM1 than Gasteiger atomic charge method is that AM1 embeds some quantum mechanic equations, which help to calculate a more accurate atomic charge than Gasteiger procedure. Mittal et al. (2009[22]) concluded that semi-empirical charges give better predictive certain 3D-QSAR models than using Gasteiger partial charge calculation methods. The second reason is that in molecular docking methods, which are based on AutoDock4 or Vina, just polar hydrogen atoms are assigned. So, partial atomic charges were not calculated for non-polar hydrogen atoms. It might affect the final SOMFA model and cause a negative deviation in computation of electrostatic contribution. The both electrostatic and shape potentials of the model C2 were computed. The contribution of electrostatic field and shape field to QSAR equation is 40 % and 70 %, respectively. Our SOMFA analysis result indicates that the shape contribution is of higher importance than the electrostatic one.
SOMFA model and HER2 active site analysis
The SOMFA shape potential for the analysis is presented as master grid in Figure 7(Fig. 7). In this figure, structure of compound 1 is shown as reference compound. Red spots indicate that steric bulk enhances activity in this region. Contrariwise, cyan spots show that steric bulk detracts from activity in this region. Favorable and unfavorable electrostatic regions are depicted in Figure 8(Fig. 8). In this figure, red spots indicate that positive charge is favored in this region. Alternatively, negative charge is disfavored in this region. Cyan spots demonstrate that negative charge is favored in this region. In other words, positive charge is disfavored in this region. Based on Figure 7(Fig. 7), ring substitution in the position R1 and R2 in compounds 1-10 can moderately affect the HER2 inhibitory potency. Compound 1 has two bulky chlorine and fluorine on its phenyl ring, which could make a hydrophobic interaction with Val 734, Thr 798, Ala 751, Leu 852 and Leu 726 (Figure 9(Fig. 9)). Since, water ID 22 acts as a portion of the active site of HER2 and it's essential for catalysis (Aertgeerts et al., 2011[1]), it was assigned in molecular docking studies. Based on Figure 9(Fig. 9), hydrogen bond is formed between compound 1 and water ID 22. The most potent compound (compound 10) has the minimum IC50. The bulky fluorobenzene moiety could interact with Leu 785, Thr 798, Val 734 and Leu 852 via hydrophobic interaction (Figure 10(Fig. 10)). These amino acids form a hydrophobic pocket that fluorobenzene moiety could lie on it. This hydrophobic moiety decreases the IC50, which is in accordance with our shape SOMFA model (Figure 7(Fig. 7)). Based on Figure 7(Fig. 7), number of carbons in the hydrocarbon tail might affect the HER2 kinase inhibitory potency. Although, compound 1 with n=1 (number of carbon in the hydroxamic tail) has an IC50 equal to 10.6, but it seems that optimal n is 5 (Cai et al., 2010[7]). Hydrocarbon tail (n=5) of hydroxamic acid lets this moiety to form a hydrogen bond with Arg 849 and Asp 863. NH of Met 801, which is an important amino acid in HER2 active site (Aertgeerts et al., 2011[1]), makes a hydrogen bond with one of the adenine moiety nitrogens. The remaining nitrogen also forms a hydrogen bond with water 22. With regard to Figure 7(Fig. 7), C-7 substitution (R3) with long chain at quinazoline ring can reduce the inhibitory activities. High IC50 of compounds 21 and 22 (478.2 for compound 22 and 225.2 for compound 22) correlate with our results (Cai et al., 2010[7]). Figure 11(Fig. 11) shows the 2-dimensional position of compound 22 in HER2 kinase active site. In spite of the fact that compound 22 contributes in several hydrogen bonds, polar Lys 753 with the positive charges in position with hydrocarbon tail might decrease the HER2 inhibitory potency. Furthermore, because of the long chain in compounds 21 and 22, conformational change of chloroflourophenyl moiety could not let this group interact with any non-polar residues via hydrophobic interaction. Our data shows that hydrophobic interaction of the moiety mentioned above could influentially increase the HER2 inhibitory potency. Table 1(Tab. 1) shows that addition of a bulky group in position R1, R2 and Y simultaneously leads to decrease in HER2 inhibitory activity. Compound 20 has chlorine in position R1 and ethyl in position Y. IC50 of this compound is 556.9. With exchange of chlorine with fluorine, IC50 was sharply decreased (compound 19). Compound 15 possesses CH2 in position Y and fluorine and chlorine in positions R1 and R2, respectively. IC50 of this compound is slightly lesser than that of compound 20. Based on our SOMFA model (Figure 7(Fig. 7)), phenyl moiety of compounds 15, 16 and 20 are located on cyan spots area. These results have a good agreement with experimentally obtained IC50 values (Cai et al., 2010[7]).
The authors suggest that based on SOMFA analysis presented in this study, more potent inhibitors of kinase domain of HER2 could be designed and synthesized.
Conclusion
In the last decade, structure-based methods have become great tools in drug design, including lead discovery and optimization. It has also been shown that structure-based methods are now able to predict, with an acceptable degree of exactness, the position of a ligand in its binding site. Combining the molecular docking and 3D-QSAR studies could be employed to design the more potent HER2 chemical inhibitors. By the aid of molecular docking, the derived 3D-QSAR models are also able to indicate which interaction sites in the binding pocket might be responsible for the variance in biological activities. Active site analysis of kinase domain of HER2 also lets us interpret and validate our QSAR model. In this study, calculated SOMFA 3D-QSAR models for quinazoline derivatives were expanded. Because of the nature of HER2 active site, which is covered by hydrophobic residues, shape contribution in our SOMFA model is a dominant factor. The number of hydrophobic interactions and hydrogen bonding between chemical inhibitors and HER2 active site could increase the inhibitory potency. These features provide some useful information that might be used in new quinazoline derivatives with a high and influential HER2 inhibitory activity.
Conflict of interest statement
The authors declare that they have no conflict of interest.
References
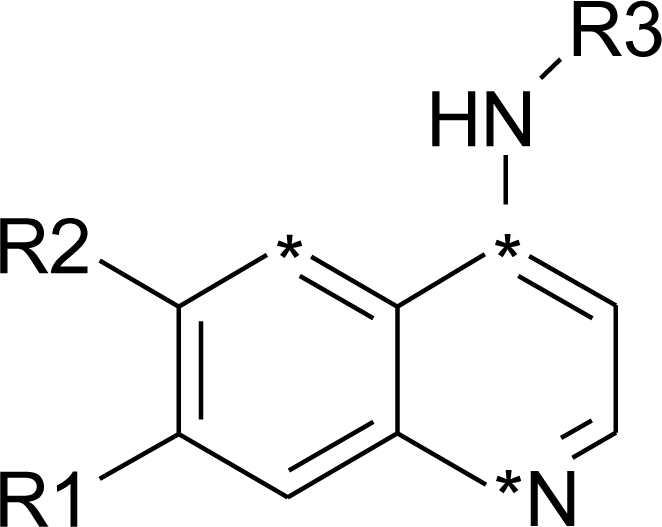
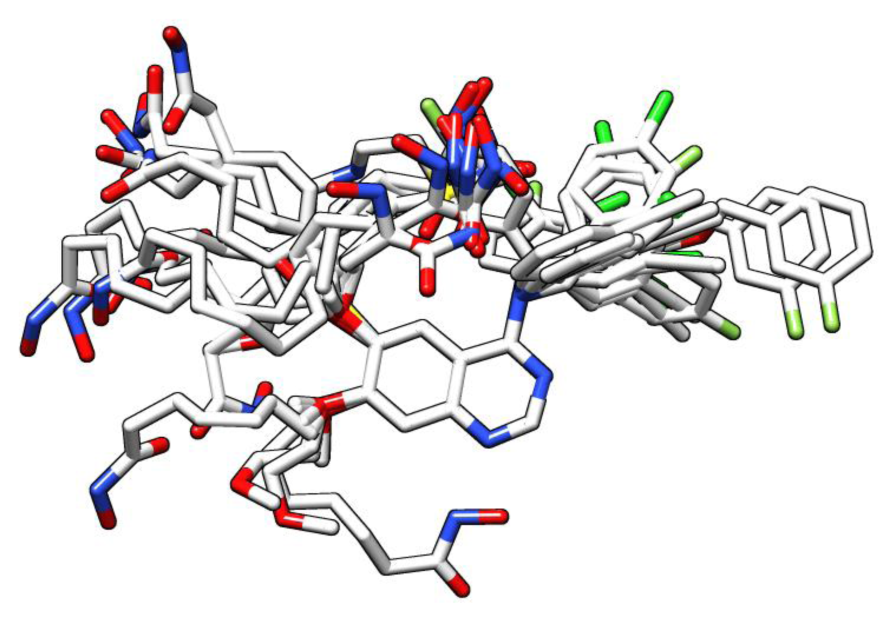
Figure 2: Superposition of all structures on reference compound. For ease of visualization, hydrogen atoms are not shown.
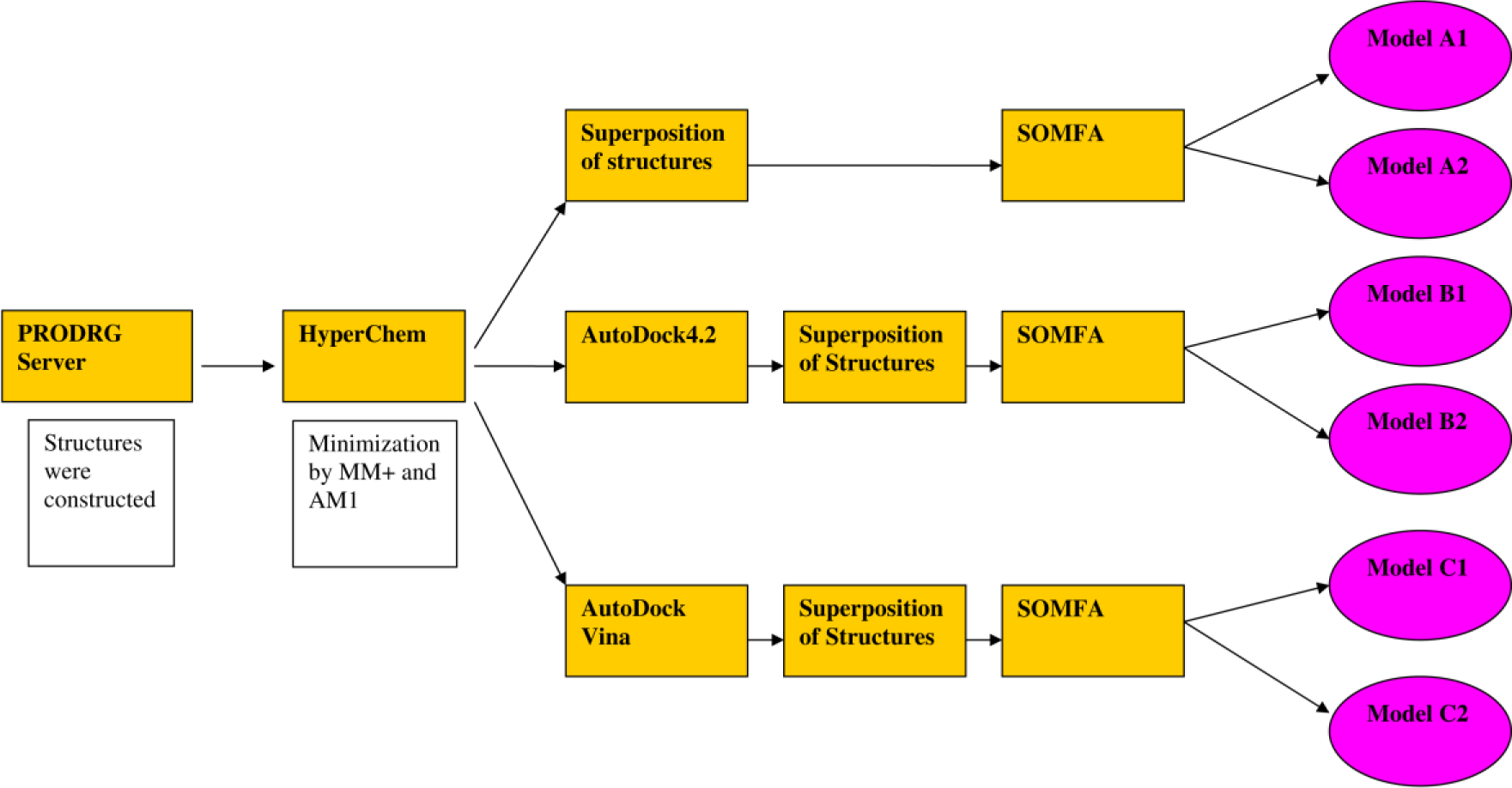
Figure 3: Flow chart of 3D-QSAR modeling procedure which is used in this study. Based on molecular docking and modeling software and SOMFA resolution utilized, for each category two models were generated. Models A1, B1 and C1 have resolution 1, whereas, resolution of 0.5 was assigned for model A2, B2 and C2.
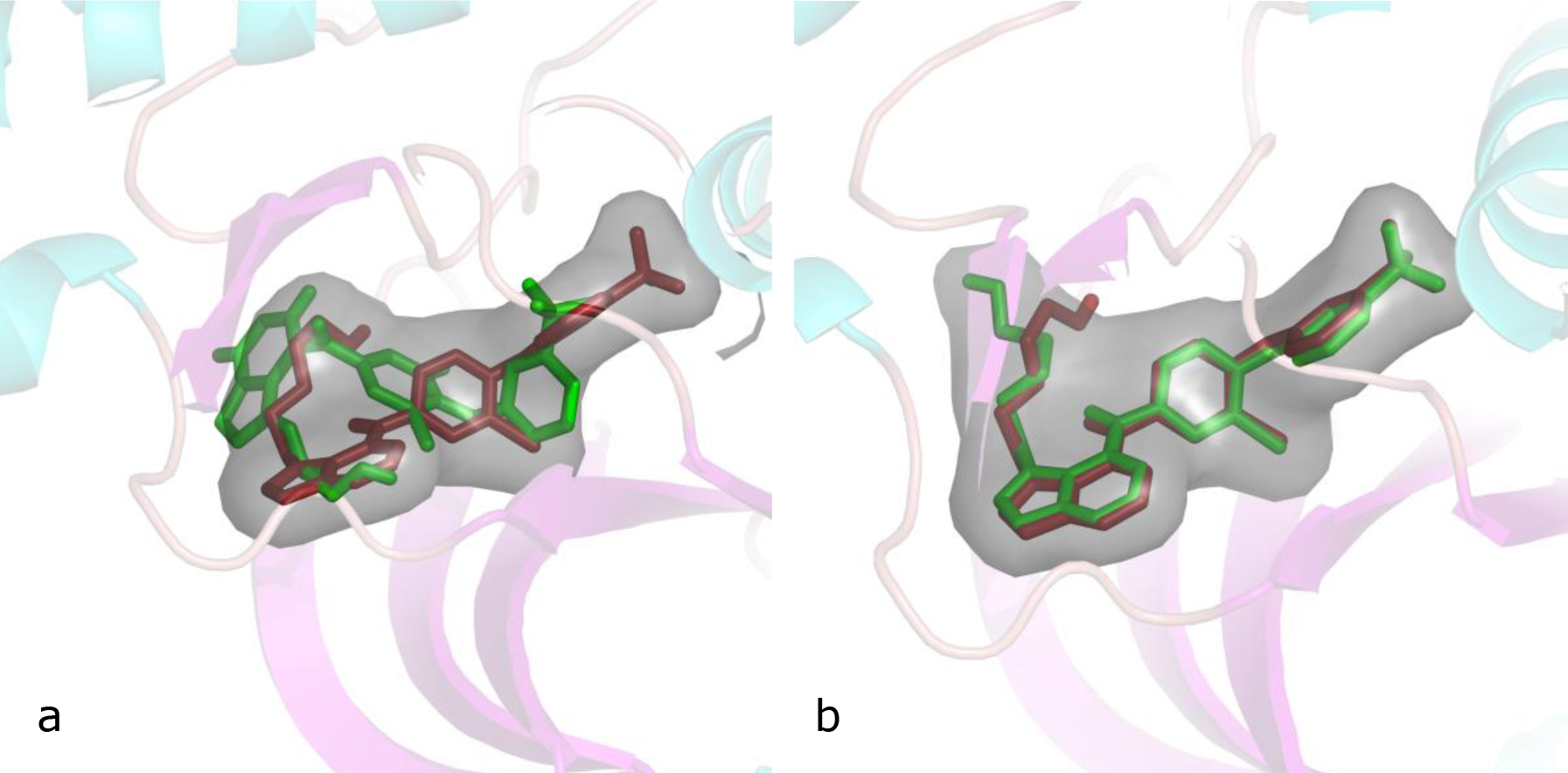
Figure 4: Cartoon view of the active pocket of kinase domain of HER2 with its ligand
(a) Superposition of the docked structure and the crystal structure of SYR127063 which is calculated by AutoDock4
(b) Superposition of the docked structure and the crystal structure of SYR127063 which is computed by AutoDock Vina. The crystal structure and docked conformation of SYR127063 are shown by red and green colors, respectively. For ease of visualization, hydrogen atoms are neglected.
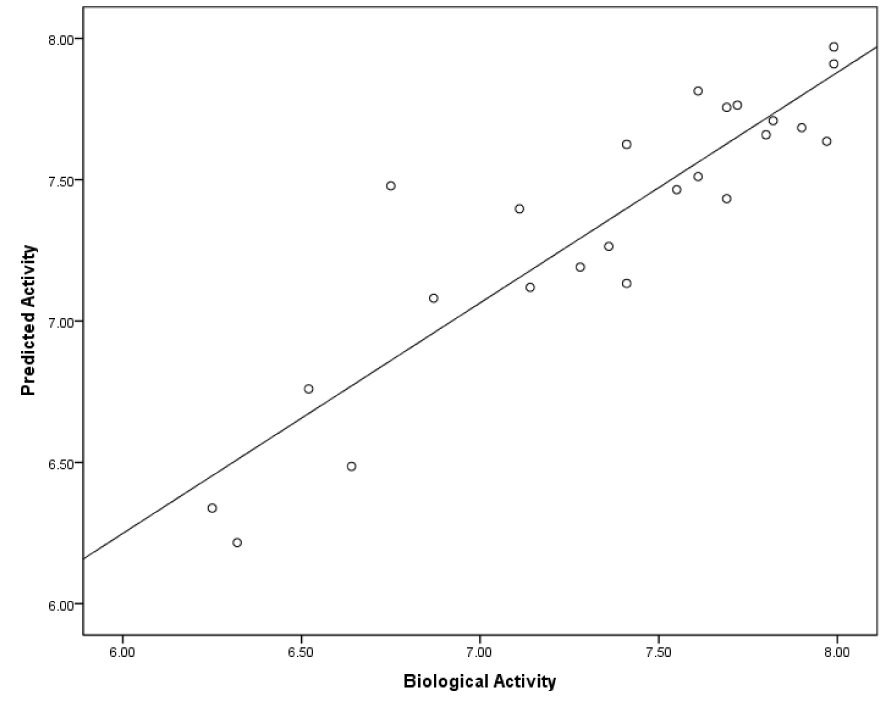
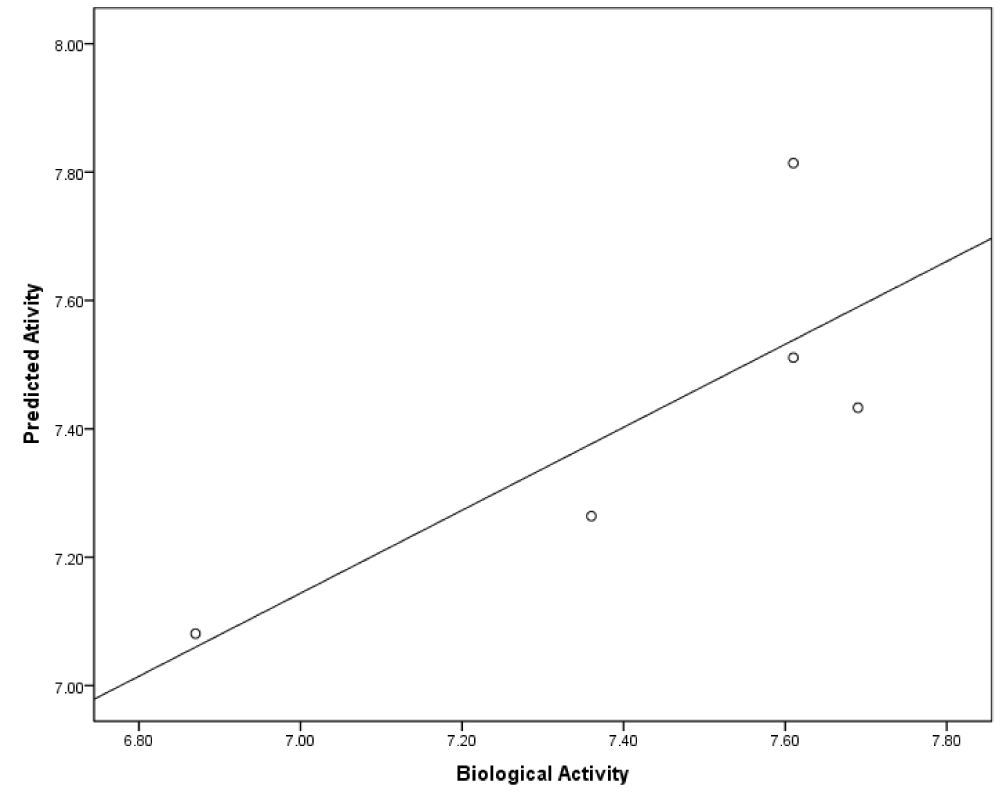
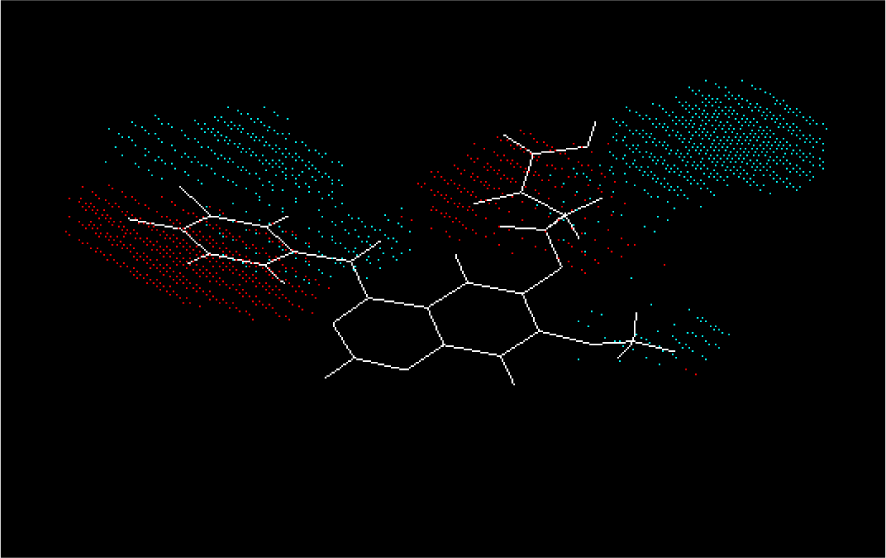
Figure 7: The shape potential master grid of model C2 with compound 1. Red spots portray areas of desirable steric interactions. Blue spots represent areas of undesirable steric interactions.
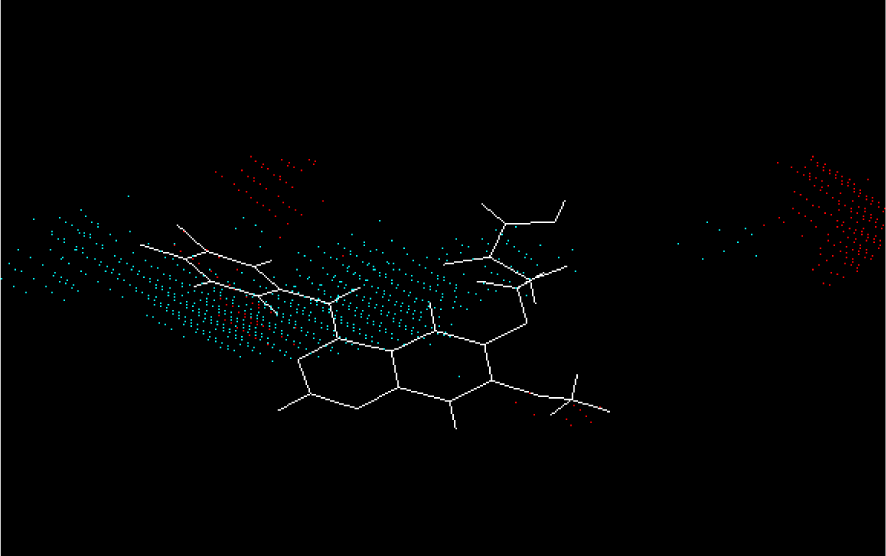
Figure 8: The electrostatic potential master grid of model C2 with compound 1. Red portrays areas where positive potential is favorable, or negative charge is unfavorable. Blue demonstrates areas where negative potential is desirable, or positive charge is undesirable.
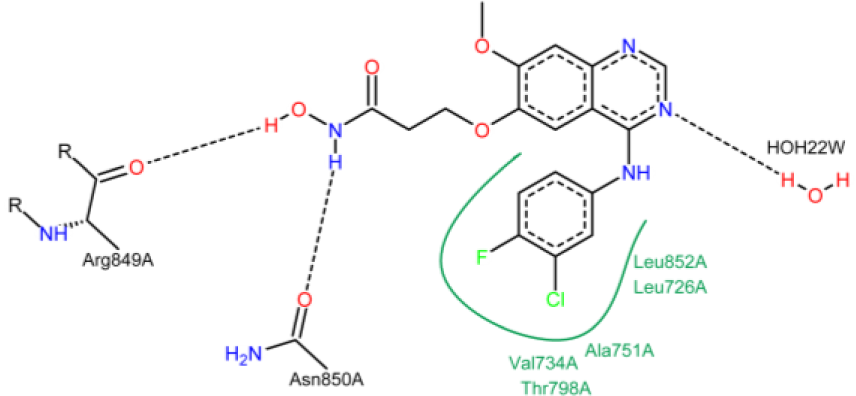
Figure 9: Schematic two-dimensional representations of the binding interactions between compound 1 and active site of HER2 kinase domain. Black dashed lines are hydrogen bonds. Hydrophobic interactions are shown by solid line green. HOH22W represents water molecule.
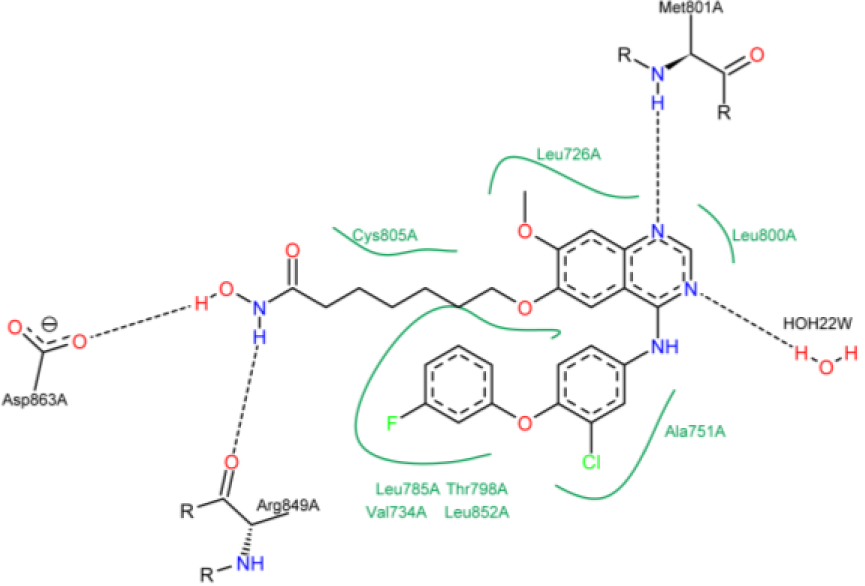
Figure 10: Schematic two-dimensional representations of the binding interactions between compound 10 and active site of HER2 kinase domain. Black dashed lines are hydrogen bonds. Hydrophobic interactions are shown by solid line green. HOH22W represents water molecule.
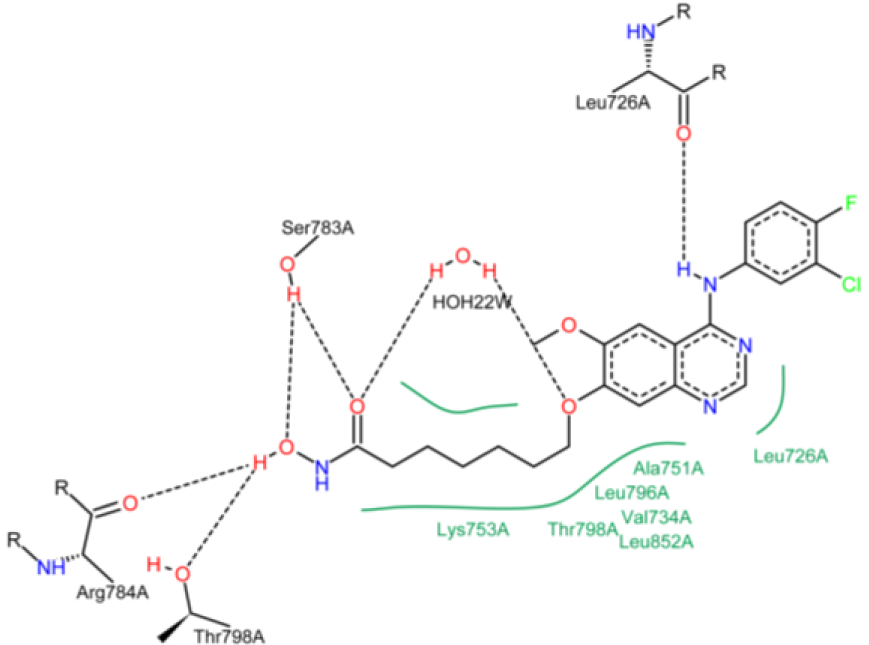
Figure 11: Schematic two-dimensional representations of the binding interactions between compound 22 and active site of HER2 kinase domain. Black dashed lines are hydrogen bonds. Hydrophobic interactions are shown by solid line green. HOH22W represents water molecule.
[*] Corresponding Author:
Sako Mirzaie, Department of Biology, Faculty of Science, Science and Research Branch, Islamic Azad University, Tehran, Iran, eMail: sako.biochem@gmail.com