Research article
Computational modeling and validation studies of 3-D structure of neuraminidase protein of H1N1 influenza A virus and subsequent in silico elucidation of piceid analogues as its potent inhibitors
Chhedi Lal Gupta1, Salman Akhtar2, Preeti Bajpaib2, K. N. Kandpal1, G. S. Desai3, Ashok K. Tiwari1[*]
1Bioinformatics Centre, Indian Veterinary Research Institute, Izatnagar–243122, India2Department of Biotechnology, Microbiology and Bioinformatics, Integral University, Lucknow-226026, India
3Division of Animal Biotechnology, Indian Veterinary Research Institute, Izatnagar-243122, India
EXCLI J 2013;12:Doc215
Abstract
Emergence of the drug resistant variants of the Influenza A virus in the recent years has aroused a great need for the development of novel neuraminidase inhibitors for controlling the pandemic. The neuraminidase (NA) protein of the influenza virus has been the most potential target for the anti-influenza. However, in the absence of any experimental structure of the drug targeting NA protein of H1N1 influenza A virus as zanamivir and oseltamivir, the comprehensive study of the interaction of the drug molecules with the target protein has been missing. Hence in this study a computational 3-D structure of neuraminidase of H1N1 influenza A virus has been developed using homology modeling technique, and the same was validated for its reliability by ProSA web server in term of energy profile & Z scores and PROCHECK program followed by Ramachandran plot. Further, the developed 3-D model had been employed for docking studies with the class of compounds as Piceid and its analogs. In this context, two novel compounds (ChemBank ID 2110359 and 3075417) were found to be more potent inhibitors of neuraminidase than control drugs as zanamivir and oseltamivir in terms of their robust binding energies, strong inhibition constant (Ki) and better hydrogen bond interactions between the protein-ligand complex. The interaction of these compounds with NA protein has been significantly studied at the molecular level.
Keywords: neuraminidase, modeler, Ramachandran plot, molecular docking, anti-influenza drugs, piceid
Introduction
Influenza virus has been a global public health concern and is responsible for significant morbidity, mortality and economic loss each year due to seasonal epidemics. The influenza pandemics occur when an influenza virus with hemagglutinin or neuraminidase against which there is very few or no preexisting immunity, emerges in the human population and is able to be transmitted efficiently from human to human (Sarkar et al., 2011[19]). These viruses belong to the Orthomyxoviridae family and can be classified into three types A, B, and C. Of these type A is clinically the most important which accounts for all of the human pandemics in the last century: the Spanish flu of 1918 (H1N1), the Asian flu in 1957 (H2N2) and the Hong Kong flu of 1968 (H3N2) (Sakudo et al., 2009[16]; Xu et al., 2008[25]).
Influenza virus can be classified by antigenic properties of two functional surface glycoproteins namely, hemagglutinin (HA) and neuraminidase (NA). 16 subtypes (H1–H16) have currently been defined for the HA protein and 9 subtypes (N1–N9) for the NA protein. NA is responsible for cleaving of sialic acid receptors from the cell surface to facilitate the elution of the progeny virions from the infected host cells (Du et al., 2010[4]). NA may also facilitate the early processing of influenza virus infection in lung epithelial cells (McKimm–Breschkin, 2000[12]). Therefore, the inhibition of influenza virus NA strongly prevents the infection from influenza virus. As importance of NA in the pathogenesis of influenza virus infection is significant, it has been the most credible target for the development of anti-influenza drugs (Lew et al., 2000[9]). Both zanamivir (Relenza) and oseltamivir (Tamiflu) are NA inhibitors and are currently used for the treatment of influenza infections (Ho et al., 2007[5]; Lew et al., 2000[9]). However, in the recent clinical practices the emergence of drug-resistant strains of influenza viruses has been reported where existing therapy for H1N1 influenza virus is not enough due to drug resistance variants (McKimm–Breschkin, 2000[12]). Thus, there has been an urgent need to designing of novel anti-influenza drug.
Natural compounds have been the mainstay of traditional medicine for thousands of years. In the current era, focus on the naturally derived compound medicines has been increasingly reported (Newman and Cragg, 2007[14]; Lam, 2007[7]). In continuation to this, here we have concentrated on the oligostibenes as an active principle component of Vitis amurensis plant, a wild growing grape distributed widely in Korea, China and Japan and which is known to show promising anti-influenza activity (Anh Nguyen et al., 2011[2]).
So far, no experimental structure of the drug-targeting NA protein of H1N1 influenza A virus is available and hence the mode and mechanism of binding of drug molecules with the NA (neuraminidase) has been an unexplored phenomenon. Resolving this fact, here we have used bioinformatics approach to model the 3-D structure of NA protein of influenza virus. Further we report the identification of some novel compounds by molecular docking studies which can inhibit the neuraminidase protein within an acceptable inhibition range, hence provide leads towards the development of anti-influenza therapy.
Materials and Methods
Sequence alignment and homology modeling
The complete amino acid sequence of neuraminidase (NA) protein of H1N1 influenza virus [Influenza A virus (A/KOL/ 2172/2009 (H1N1))] was retrieved from the NCBI protein database (accession no. ADJ18735.1). BLAST-P program against the protein data bank was performed to retrieve suitable templates for homology modeling (Altschul et al., 1997[1]). The atomic coordinates of crystal structure of native 1918 H1N1 neuraminidase (PDB ID: 3CYE) was taken as template to generate the 3-D model of NA retrieved from the Protein Data Bank (http://www.rcsb.org/pdb). The sequence alignment among target and template was carried out using CLUSTALW program (Thompson et al., 1994[22]).
The initial model of 3-D structure of NA protein of H1N1 influenza A virus was built by Homology Modeling method using Modeller9v9 on window based operating system (Sali and Blundell, 1993[17]); a program for comparative protein structure modeling optimally satisfying spatial restraints derived from the alignment and expressed as Probability Density Functions (pdfs) for the features restrained which includes: (i) Homology derived restraints on the distances and dihedral angles in the target sequence extracted from its alignment with template structure. (ii) Stereochemical restraints such as bond length and bond angles preferences obtained from the CHARMM22 molecular mechanics force field (MacKerell et al., 1998[11]). (iii) statistical preferences for dihedral angles and non-bonded inter atomic distances, from a representative set of known protein structures (Sali and Overington, 1994[18]) and (iv) optional manually curated restraints, such as those from NMR spectroscopy, rules of secondary structure packing, cross linking experiments fluorescence spectroscopy, image reconstruction from electron microscopy, side directed mutagenesis and intuition. The pdfs restrain Cα-Cα distances, main chain N-O and main chain-side chain dihedral angles. The 3-D model of protein is obtained by optimization of molecular pdf such that the model violates the input restraints as little as possible. The molecular pdf is derived as combination of pdfs restraining individual spatial feature of the whole molecule. The optimization procedure is variable target function method that applies the conjugate gradients algorithm to positions of all non hydrogen atoms. Modeller generated several preliminary models by the satisfaction of spatial restraints which were ranked based on their DOPE (Discrete Optimized Potential Energy) scores. Models with the lowest DOPE score were selected in order to check the stereo chemical quality of the given protein structure, as compared with well refined structures at the same resolution and to give indication of its local, residue by residue reliability by PROCHECK (http://nihserver.mbi.ucla.edu/SAVES). The model with maximum number of residues in core region in the Ramachandran plot was selected for further studies. Finally energy minimization of the constructed structure was performed until the energy gradient was lower than 0.1 Kcal/mole Å using CharMM force field.
Model validation
The modeled 3-D structure of NA was validated by PROCHECK program which generates Ramachandran plot of amino acid sequences in allowed and disallowed regions (Laskowski et al., 1993[8]). The ProSA web server was employed to evaluate energy profile and verify the structure in term of Z score, representing the overall quality and measures the deviation of total energy (Wiederstein and Sippl, 2007[24]).
Docking simulations
Piceid and its analogues were retrieved from ChemBank database (http://chembank.broadinstitute.org) for molecular docking studies with the modeled NA protein. The compounds were downloaded in sdf format from ChemBank database and converted into pdb format by OpenBabel tool. The compounds were energy minimized using Chimera (Pettersen et al., 2004[15]). Gasteiger charges were applied to them and were subjected to single step minimization using steepest descent method for 500 steps at step size of 0.02 with updated interval of 10. The molecular docking was performed using AutoDock Tools 4.0 in order to find the preferred binding conformations of the ligands in the receptor (Morris et al., 1998[13]). The analysis of the binding conformation of protein-ligand complex was performed using a scoring function based on the free energy of binding (Huey et al., 2007[6]). Among the stochastic search algorithms offered by AutoDock suite, the Lamarckian Genetic Algorithm (LGA) which combines global search (Genetic Algorithm alone) to local search (Solis and Wets algorithm) was chosen (Solis and Wets, 1981[20]). The grid parameter file of receptor was generated using AutoDock 4.0. The number of grid points in x, y, and z-axes were 60×60×60 Å. The distance between two connecting grid points was 0.375 Å. Lamarckian Genetic Algorithm (LGA) was used for docking calculations. 15 search attempts (GA_run parameter) and population size of 250 were performed for ligand. The maximum number of energy evaluations before the termination of LGA run was 2500000 and the maximum number of generations was 27000 and other parameters were set to the software's default values.
Results and Discussion
Sequence analysis and homology model analysis
The neuraminidase (NA) protein is found localized on the viral surface and subsequently arrests the release of new virions from the infected cell to neighbouring cells. This protein is 378 amino acids long with estimated molecular weight of 41.7 kDa. The similarity searching results in our study reveals that it has 88 % identity and 93 % similarity with the crystal structure of 1918 H1N1 neuraminidase (3CYE), which can be significantly used an excellent template for modeling of NA protein. Owing to this, both the amino acid sequences of target (NA) and template (1918 H1N1 neuraminidase) were aligned (Figure 1(Fig. 1)) with the formation of an alignment file. Modeler program was successfully able to generate a 3D model of NA protein by the satisfaction of spatial restraints using the crystal structure 3CYE. A total of five models was generated by modeler and one of which was selected as a model of target NA protein.
Furthermore, the validation studies of the generated model of NA using PROCHECK program showed 89.8 % residues in most favored regions, 9.9 % in additionally allowed regions and 0.3 % in generously allowed regions whereas 0.0 % of amino acid residues were found in the disallowed region and promisingly the overall G-Factor was -0.19 (Table 1(Tab. 1) and Figure 2(Fig. 2)). The model represented good ERRAT plot with the overall quality factor of 77.68.
The superimpositioning of NA model with the template protein 1918 H1N1 influenza virus neuraminidase as shown in Figure 3(Fig. 3) further revealed that the developed model is almost identical as that to our template. Also the superimposition indicated that their root mean square deviation (RMSD) value was 0.151 Å, which are the characteristics of a good overall structural alignment. The above results clearly suggest out that the developed model is promising and reliable.
The Z score of the protein under study is displayed in the plot with a dark black point (Figure 4(Fig. 4)). The Z score value of the obtained model of NA is −5.12, which is within the acceptable range −10 to 10 and is located within the space of protein related to X-ray (Figure 4(Fig. 4)). This is very close with the Z score value of the template (−5.19) suggesting that the obtained model is reliable and closes to experimentally determined structure. It has been reported that the Z score is dependent on the length of the protein and negative Z-scores are very good for a reliable model. The Z score represents the overall quality and measures the deviation of the total energy of the protein structure (Yadav et al., 2012[26]).
The quality of the protein folds of NA model was also evaluated in terms of energy function of amino acid residues. In general, folding energy of the protein showed minimum value as this accounts for the stability and nativity of the molecules. The energy profile of NA model in comparison to that of the X-ray structure of template is presented in Figure 5(Fig. 5). The trend of the variation of the protein folding energy in NA model was in good agreement with that of the X-ray structure of template (3CYE).
The obtained results clearly propose the developed model of the neuraminidase (NA) protein filtered through various parameters of validation and authenticity to be significant and reliable for carrying out further studies.
Identification of novel inhibitors of NA through molecular docking analysis
A selected list of Piceid analogs along with the two control drugs zanamivir and oseltamivir available in ChemBank database were docked into NA structure using AutoDock Tools (Table 2(Tab. 2)).
The docking studies indicated that two novel piceid compounds (ChemBank ID 2110359 and 3075417) (Figure 6a and 6b(Fig. 6)) produced better binding energy and strong inhibition constant among other compounds considered for this study. The binding energy of these [3-hydroxy-5-[(E)-2-(4-hydroxyphenyl)vinyl]phenyl beta-D-glucopyranoside (2110359)] and [rhapontin (3075417)] compounds were -8.02 kcal/mol and -8.23 kcal/mol with estimated inhibition constant of 1.31 µm and 0.931 µm respectively. Even their binding energies were found better than that of control drugs zanamivir and oseltamivir with just -5.46 kcal/mol and -4.86 kcal/mol respectively. Both these novel compounds also fulfill the criterion of Lipinski's rule of five with just one violation (Supplementary materialSupplementary material.pdf). Therefore these two novel compounds were selected for further comparative study with commonly used anti-influenza drugs zanamivir and oseltamivir.
Understanding the complex molecular binding processes is undoubtedly of critical importance in structure-based drug design, and much effort is being invested in experimental and computational methods to resolve binding (Buch et al., 2011[3]). The electrostatic, Van der Waals, hydrogen bonding, hydrophobic interactions are the driving force behind the formation of molecular complex (Liang et al., 2009[10]). The ligand binding mechanism study of selected novel compounds (2110359 and 3075417) with control drugs zanamivir and oseltamivir (Figure 6c and 6d(Fig. 6)) carried out by molecular docking analysis showed that both the novel compounds and control drugs bound to neuraminidase with strong interaction energies and formed hydrogen bond with NA.
To identify the binding forces contributing to NA-compound bound complexes, the Internal energy, the addition of Van der Waals, hydrogen bond and desolvation energy (vdw + Hbond + desolv energy), electrostatic energy and total intermolecular energy of both novel compounds and control drugs with NA were also calculated (Table 3(Tab. 3)).
In this analysis the total intermolecular energy of two novel compounds 2110359 and 3075417 (-8.83 Kcal/mol and -9.50 Kcal/mol) was found lower than control drugs zanamivir and oseltamivir (-4.45 Kcal/mol and -5.99 Kcal/mol), stating that the identified novel compound had better potency.
Hydrogen bonds formed between compound and protein usually contribute to the stability of the protein-ligand complexes, and large number of hydrogen bonds form more stable complex (Steiner and Koellner, 2001[21]; Weiss et al., 2001[23]). Our results also showed that both the novel compounds formed a large number of hydrogen bonds (2110359, five hydrogen bonds and 3075417, six hydrogen bonds) with NA (Figure 7a, b(Fig. 7)), while the control drugs zanamivir formed four and oseltamivir formed three hydrogen bonds respectively (Figure 7c, d(Fig. 7)). Hydrogen bonding interaction study with these novel compounds revealed that active residues of NA were also involved in the formation of hydrogen bond, suggesting the screened novel compounds forms more stable complex than the two anti-influenza drugs in use (Table 4(Tab. 4)). Therefore, in the study of ligand binding mechanism, we consider that the novel compounds (2110359 and 3075417) have stronger interaction energies with NA.
From the comparative study it has been noticed that the novel compounds (2110359 and 3075417) have a strong tendency to be used as an anti-influenza agent by the inhibition of NA protein. Therefore, the related biological experiments should also be tested besides the neuraminidase inhibition in silico experiments.
Our study has successfully predicted a reliable computational 3-D model for the neuraminidase NA protein, the most targeted protein in the anti-influenza therapy. PROCHECK, ERRAT and ProSA web servers have further validated the developed neuraminidase model. Molecular docking studies carried out to screen novel compounds for NA inhibition have indicated the compounds 2110359 and 3075417 as effective inhibitors with robust binding energy, strong hydrogen bond interactions than the control drugs zanamivir and oseltamivir. The study foresees the 3-D structures of NA and it complexes with these compounds (2110359 and 3075417) as novel insights in the form of inhibitory action and may provide basis for development of new anti-influenza drugs.
Acknowledgement
We are thankful to the Director, Indian Veterinary Research Institute, Izatnagar for providing the infrastructural facilities and Department of Biotechnology, New Delhi for the support of this investigation.
References
File-Attachments
- Supplementary material.pdf (148,32 KB)
Supplementary material

Figure 1: Alignment of NA with template 3CYE. Star (*) indicated identical amino acids, colon (:) for similar amino acids and dot (.) for nearly similar ones
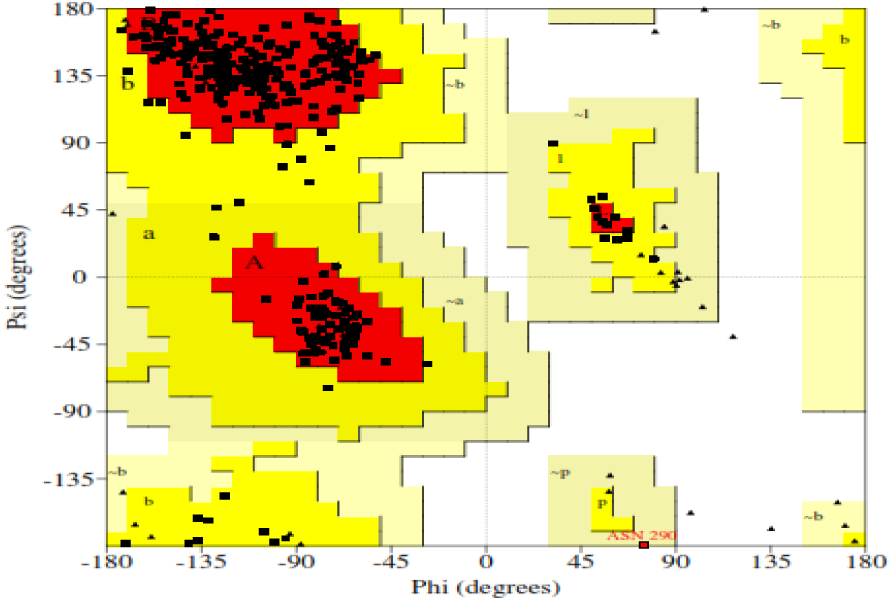
Figure 2: Ramachandran plot of NA model showing 89.8 % of amino acid residues in core region (red color)
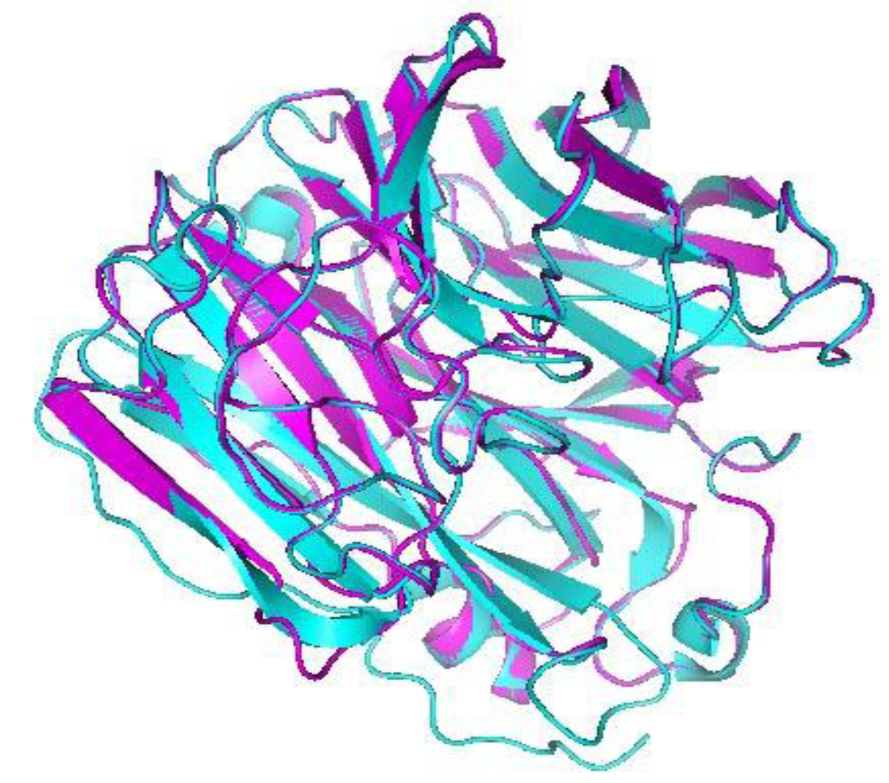
Figure 3: Superposition of NA model with its template protein 1918 H1N1 influenza virus neuraminidase. Magenta ribbon representation of NA model and Cyan ribbon representation of 1918 H1N1 influenza virus neuraminidase. Their root mean square deviation (RMSD) value was 0.151 Å.
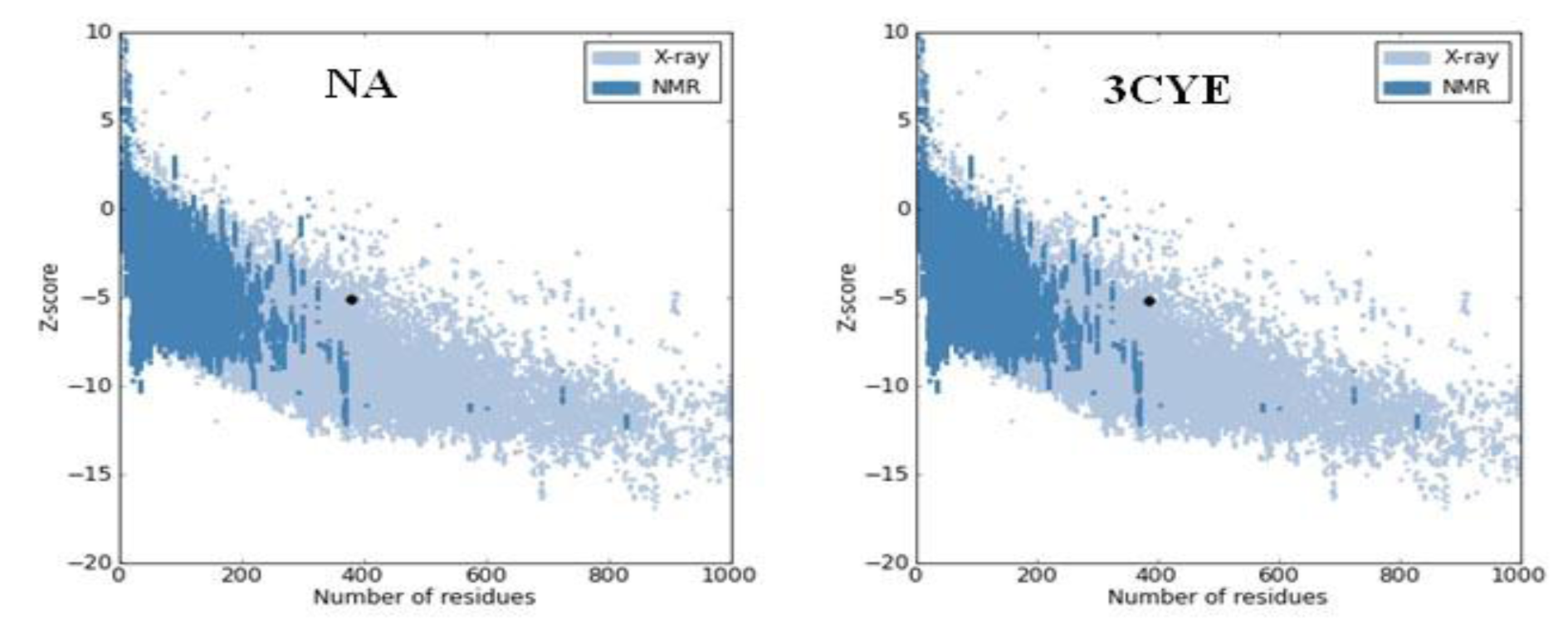
Figure 4: The Z score plot represents Z score value of proteins. The two dark black points represent Z score of the NA model and template protein (3CYE).
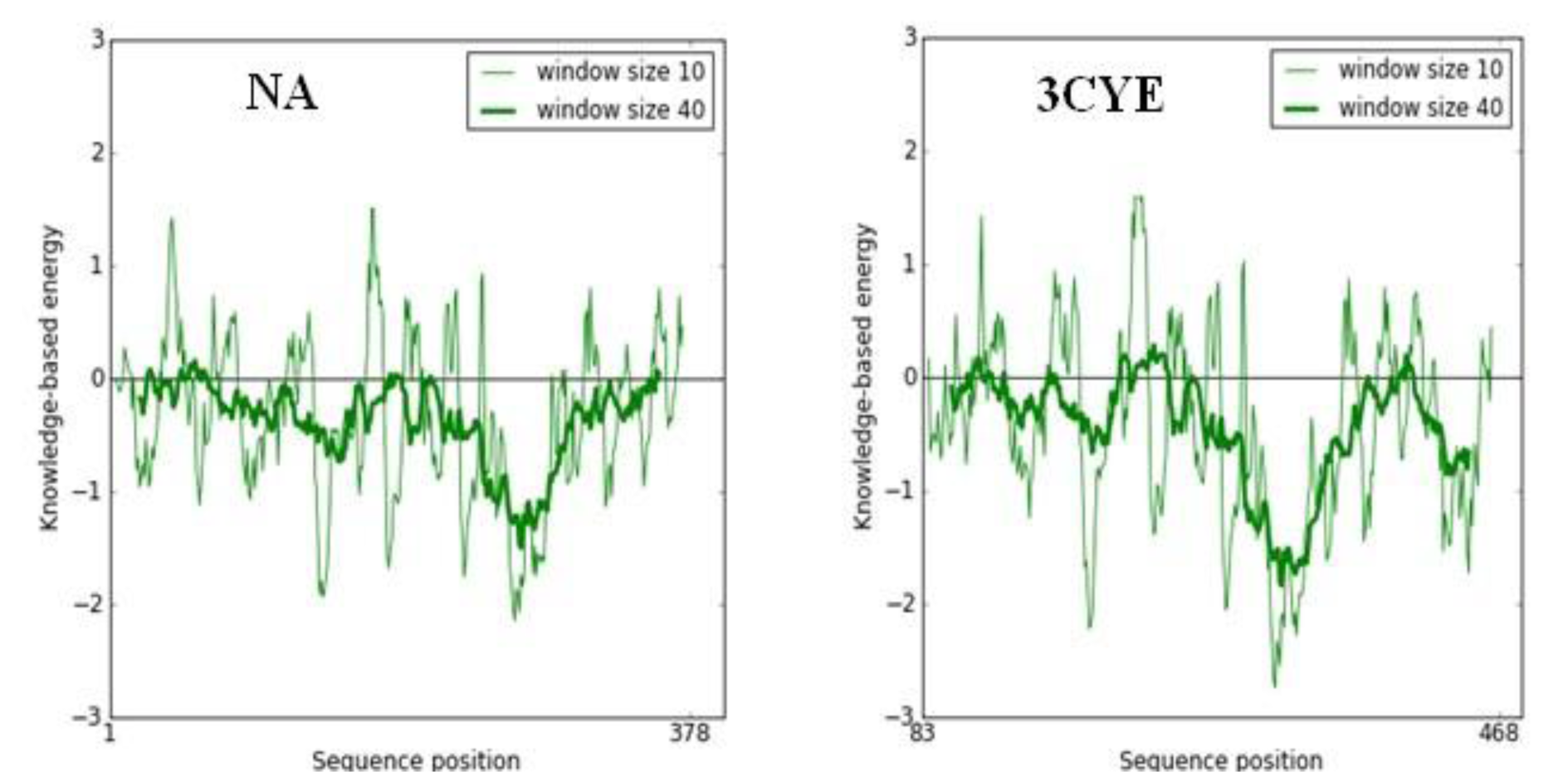
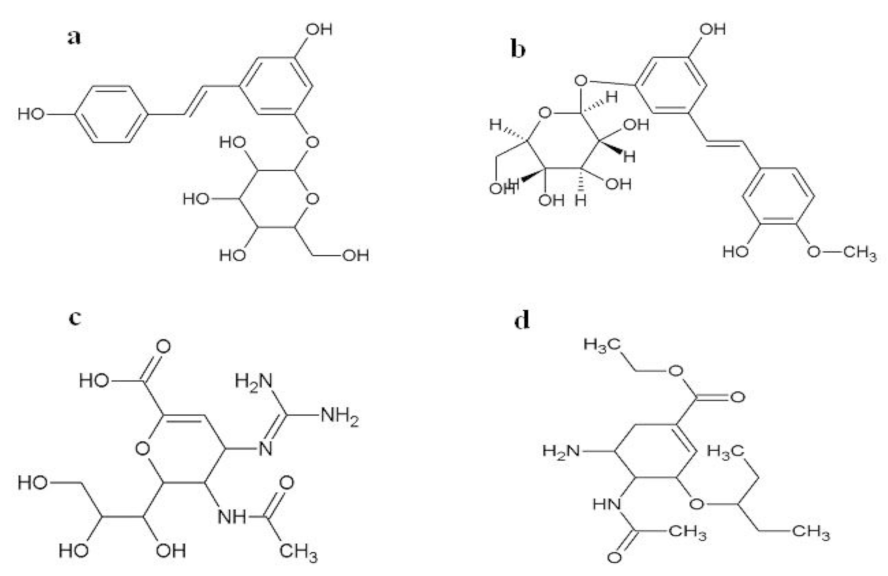
Figure 6: The 2D chemical structure of two novel compounds showing good binding energies (a) 2110359, (b) 3075417 and the anti-influenza drugs (c) zanamivir, (d) oseltamivir.
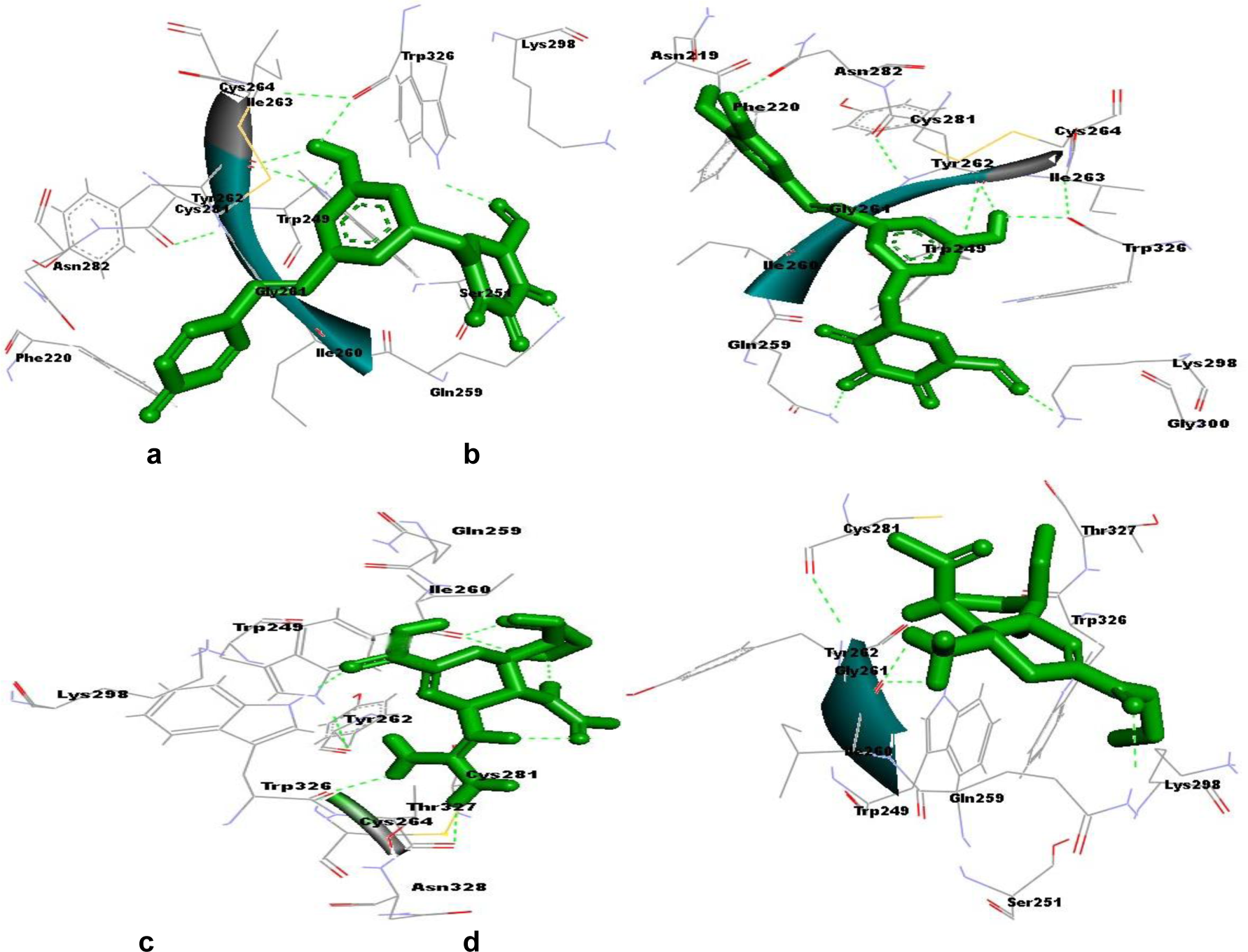
Figure 7: a: Surrounding residues and hydrogen bonds formed between compound 2110359 (in green color) and neuraminidase. b: Surrounding residues and hydrogen bonds formed between compound 3075417 (in green color) and neuraminidase. c: Surrounding residues and hydrogen bonds formed between zanamivir (in green color) and neuraminidase. d: Surrounding residues and hydrogen bonds formed between oseltamivir (in green color) and neuraminidase. Hydrogen bonds are shown by green dash line.
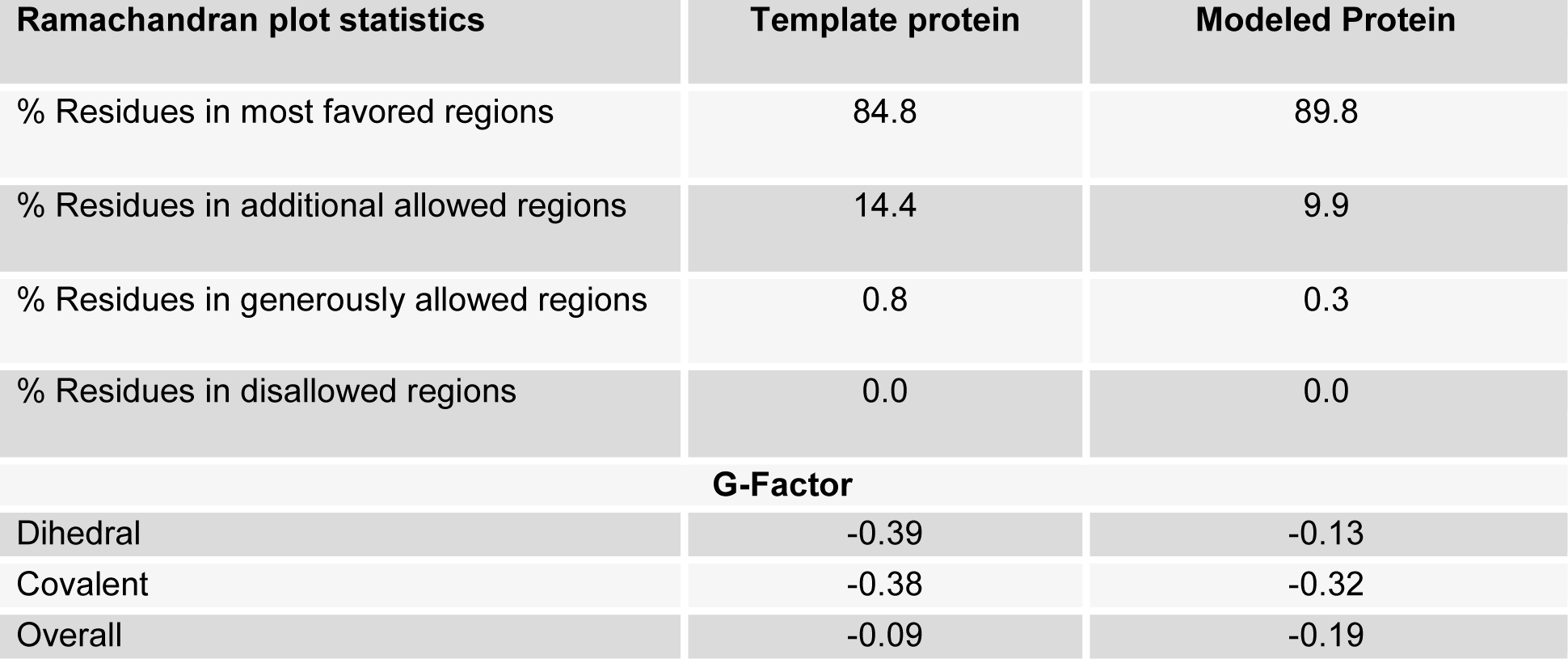
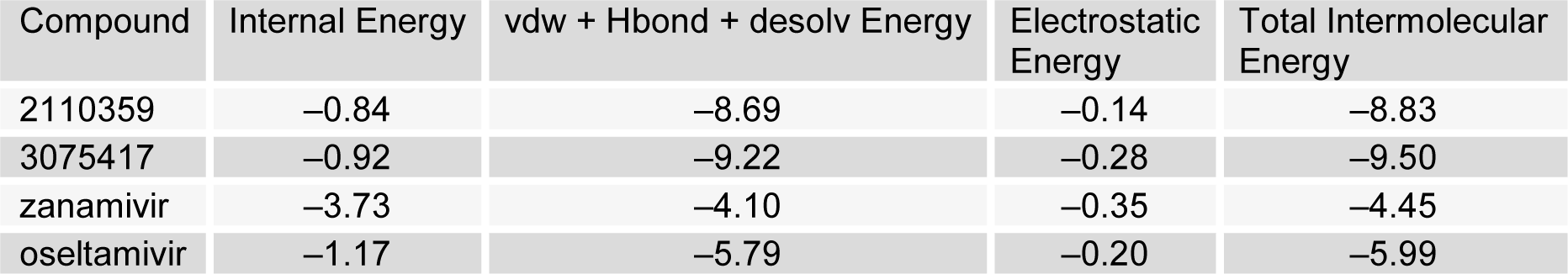
Table 3: Interaction energies of compound 2110359, 3075417, zanamivir and oseltamivir with NA (Kcal/mol) obtained from molecular docking
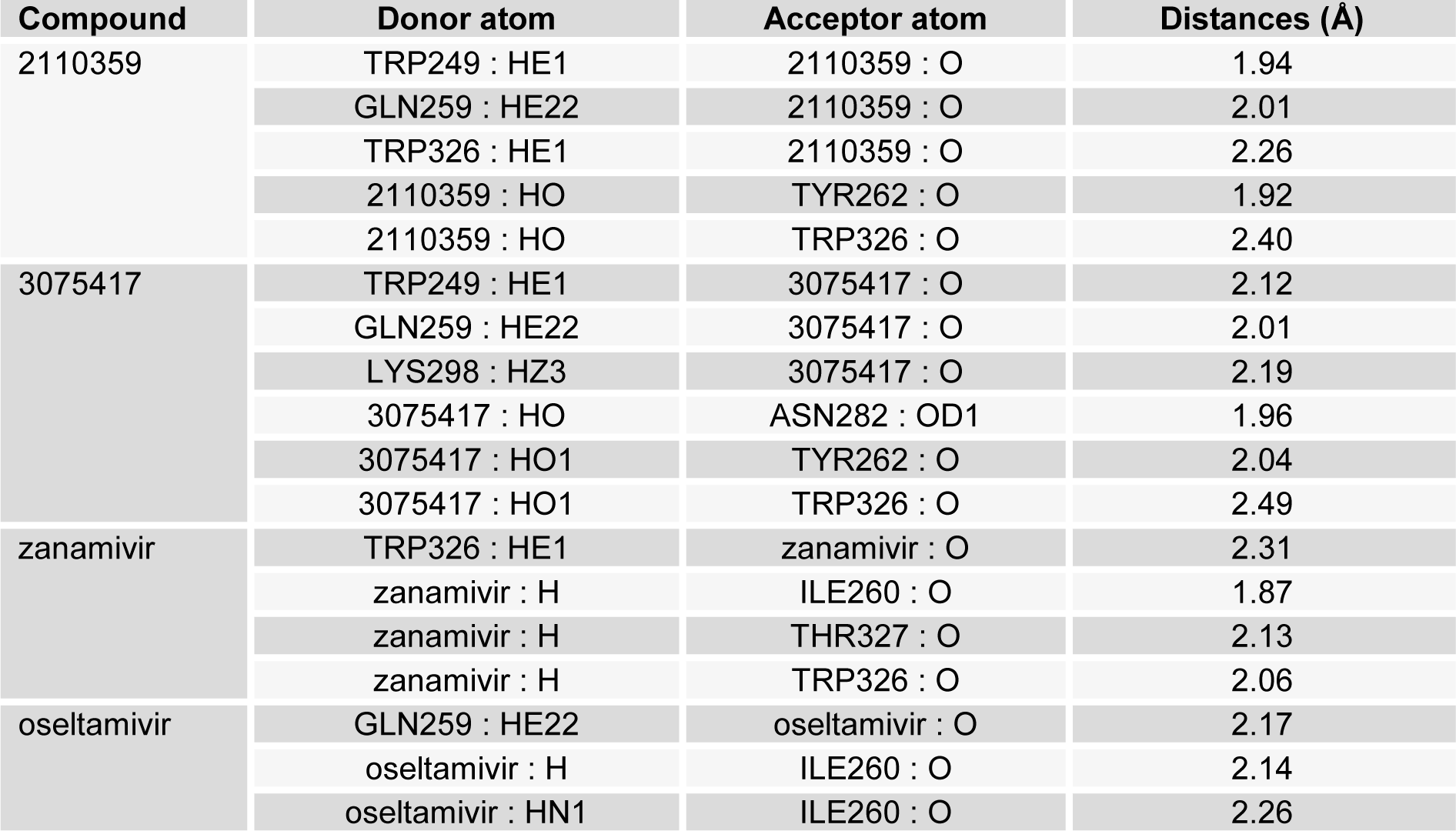
Table 4: Hydrogen bond interaction studies of compound 2110359, 3075417, zanamivir and oseltamivir with NA
[*] Corresponding Author:
Ashok K. Tiwari, Bioinformatics Centre, Indian Veterinary Research Institute, Izatnagar–243122, India, Mobile: +91– 9457257425, eMail: ashokktiwari63@gmail.com, aktiwari63@yahoo.com