Research article
Acceleration of pro-caspase-3 maturation and cell migration inhibition in human breast cancer cells by phytoconstituents of Rheum emodi rhizome extracts
D.R. Naveen Kumar1, V. Cijo George1, P.K. Suresh1, R. Ashok Kumar2[*]
1School of Bio Sciences and Technology, VIT University, Vellore - 632 014, India2Department of Zoology, Government Arts College, Dharmapuri - 636 705, India
EXCLI J 2013;12:Doc462
Abstract
The aggressive nature of estrogen receptor (ER)-negative breast cancer subtype obligates for innovative targeted therapies. The present study aimed to investigate the phytoconstituents and specific anticancer activities of Rheum emodi rhizome, a known food source used locally to treat various ailments. Petroleum ether extracts (hot [PHR] and cold [PCR]) of R. emodi, exhibited significant free radical scavenging potentials through DPPH and reducing power assays, rendering them as good sources of antioxidants. The extracts, PHR and PCR had shown significant (P < 0.05) cancer-cell-specific cytotoxicity in the assayed cells (MDA-MB-231 [breast carcinoma] and WRL-68 [non-tumoral]) at 100 μg/ml, and 50 and 100 μg/ml concentrations respectively. Extracts also induced fervent apoptosis in ER-negative cells (MDA-MB-231) compared to ER-positive subtype (MCF-7), and found to involve CPP32/caspase-3 in its apoptosis induction mechanism. Moreover, extracts had an inevitable potential to inhibit the migration of metastatic breast cancer cells (MDA-MB-231) in vitro. Further, the active principles of extracts were identified through HPLC and GC-MS analysis to reveal major polyphenolics, 4,7-Dimethyl-(octahydro)indolo[4,3-fg]quinolin-10-one, 5-Oxo-isolongifolene, Valencene-2, and other quinone, quinoline and anthraquinone derivatives. The extracts are thus good candidates to target malignant ER-negative breast cancer, and the identified phytoconstituents could be eluted in further exploratory studies for use in dietary-based anti-breast cancer therapies.
Keywords: cancer-specific cytotoxicity, ER-negative breast cancer, caspase-3, anti-metastasis, HPLC, GC-MS
Introduction
Cancer is one of the largest causes of mortality in human race. Of which, breast cancer is the most common in women, and standing second as leading causes of death (CDC, 2012[11]). Regardless of advances in treatment, resistance development to cancer drugs is a major hindrance in treating malignant cancers. The nature of heterogeneity and the ability to metastasize necessitates the need for innovative breast cancer therapies. Predominantly, the modern therapies are expected to explicitly target on specific genes or their proteins in comparison to conventional cytotoxic chemotherapy (Rochefort et al., 2003[44]). The targeted therapies are also anticipated to possess higher efficiency with lesser non-specific toxicity in patients (Gibbs, 2000[24]). Being the first successful hormonal therapy in breast cancer, anti-estrogen therapy is limited to ER-positive subtype (Hsieh et al., 2011[26]). Nevertheless, ER-negative breast cancers are reported to be the most aggressive subtype (Putti et al., 2005[40]), requiring the urgent attention for additional targeted therapies (Rochefort et al., 2003[44]; Hsieh et al., 2011[26]). In most of the cancer types, the course of carcinogenesis is a multi-step progress that includes initiation, promotion, progression and metastasis of tumor.
Copious phytochemicals are being reported to arrest or inhibit various stages of carcinogenesis in breast cancer. The targets of such chemopreventive agents vary systematically as, prevention of cancer initiation through DNA repair, free-radical scavenging, detoxification and blocking carcinogen metabolism, treating promotion and progression by inhibition of cell proliferation, migration and angiogenesis, apoptosis induction, and cancer cell-differentiation and cell death (Shu et al., 2010[47]). Of these, the therapy involving cell cycle arrest and/or apoptosis induction is considered as one of the promising strategies for chemoprevention (Pan and Ho, 2008[37]). Apoptosis induction is attained by both intrinsic (mitochondrial) and extrinsic (death receptor) mechanisms. Conversely, both the pathways converge and progress through a common effector caspase; caspase-3, which results in immediate cell death through cellular targets (Janicke et al., 1998[28]). A potent chemopreventive agent also entails inhibition of cancer cell migration in advanced stages of malignancy (metastasis), reported to cause major mortality in cancer (Fidler, 2003[22]). Hence, targeting by an agent with caspase-3 activation specificity could be a successful strategy in treating breast cancer malignancy (Putt et al., 2006[39]).
Since decades, plant sources are being the promising candidates for their higher efficacies as chemopreventives. Obstinate evidences have been reported for an association of regular consumption of plant products as dietary supplements and reduced risk of cancer, further, whose screening in vitro could provide imperative data for selection of plant extracts with potential antineoplastic properties for exploratory studies (Luo et al., 2011[33]; Sreelatha et al., 2011[49]). Rheum emodi Wall. ex Meissn. (Polygonacea) is one such well known Himalayan herb, which has beforehand reports for its use in Ayurvedic and traditional medicine systems to treat various ailments (Singh et al., 2010[48]), and was recorded as an important source of food for tribal and rural people of Kashmir (Kounsar et al., 2011[30]). The plant was previously showed to possess few well known therapeutic compounds such as chrysophanol, physcion and emodin (Babu et al., 2003[7]). R. emodi has also been previously reported for its antioxidant, cytotoxic and apoptotic properties (Babu et al., 2003[7]; Rajkumar et al., 2010[41]; 2011[42]). However, there is a lack of reports pertaining to the mechanism of action, and other phytoconstituents responsible for the stated bioactivities of these extracts. This created a scientific state of interest to explore the overseen components of R. emodi, which was the primary focus of this study, where the petroleum ether extracts of R. emodi have been scrutinized for their mode of action and their phytochemical constituents with promising and specific anticancer activities.
Materials and Methods
Chemicals
2,2-Diphenyl-1-picrylhydrazyl (DPPH), ascorbic acid, gallic acid, phenazine methosulfate (PMS) (also known as N-methylphenazonium methosulfate), L-15 (Leibovitz) cell culture medium (with L-glutamine) and MEM (minimal essential medium) cell culture medium (with Earle's salt, NEAA and L-glutamine) were purchased from Himedia Laboratories Pvt. Ltd. (India). XTT {2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino) carbonyl]-2H-tetrazolium hydroxide} was obtained from Sigma Chemical Co. (St. Louis, MO, USA). Folin-Ciocalteau reagent was procured from Sisco Research Lab (India). Cellular DNA fragmentation ELISA (Cat. No. 11 585 045 001) to determine apoptosis was procured from Roche Diagnostics, Germany. Caspase-3/CPP32 Colorimetric Assay Kit (Cat. No. K106-25) was procured from BioVision Incorporated, CA, USA. The remaining chemicals and solvents used were of standard analytical grade and HPLC grade, respectively.
Plant material
Rheum emodi rhizomes was collected from their natural habitat in the Garhwal Himalayas at Chamoli (30º 24' N, 79º 21' E), Uttaranchal, India. The specimens collected were shade-dried, powdered and used for solvent extraction. A voucher specimen was maintained at our laboratory for future reference (Accession no.: VIT/SBCBE/CCL/07/6/04; Dated: June 11, 2007).
Extraction
Rhizome powder was extracted with petroleum ether by two experimental schemes. Petroleum ether hot extract (PHR) of the rhizome powder was obtained using a Soxhlet apparatus [with the powder (g): solvent (ml) ratio of 1:6] and the petroleum ether cold extract (PCR) was obtained by the method of maceration at room temperature with the flask shaken at regular intervals. Further, the two extracts were evaporated to dryness at 40 ºC under reduced pressure [petroleum ether: 180 mbar in a rotary evaporator (Büchi, Switzerland)]. The samples were then stored in a vacuum desiccator at room temperature until further use.
Estimation of antioxidant potentials
Reducing power assay
The reducing power of the extracts was analysed by its ability to reduce Fe+3- Fe+2 according to the method described by Yildirim et al. (2001[55]). The extracts (20, 40, 60, 80 and 100 μg) were added with 2.5 mL of 0.2 M phosphate buffer (pH 6.6; 1.79 % NaH2PO4 and 1.89 % Na2HPO4) and 2.5 mL of 1 % potassium ferricyanide. The mixture was incubated at 50 ºC for 30 min, after which 2.5 ml of trichloroacetic acid (10 % solution) was added to the tubes and centrifuged at 3000 rpm for 10 min. The upper layer solution (2.5 ml) was taken and added with an equal volume of distilled water and 0.5 mL of 0.1 % ferric chloride. Absorbance was then read at 700 nm using Cary 50 UV-Vis spectrophotometer (Varian, Inc., CA, USA). Increasing absorbance value of the reaction mixture indicates an increase in reducing power of the extracts.
DPPH radical scavenging assay
The DPPH• radical scavenging ability of the extracts was measured by the method of Blois (1958[9]) with some modifications. Varying concentrations (200, 400, 600, 800 and 1000 µg) of extracts were taken in separate test tubes and made up to 0.5 ml using ethanol. DPPH● solution, 3ml (0.1 mM in ethanol), was added to all the tubes. The tube with ethanol and DPPH● solution alone was maintained as controls. The tubes were incubated in the dark for 30 min and the absorbance was measured at 517 nm with ethanol as blank. The percentage radical scavenging (RS %) was calculated using the formula:
RS % = [(Ac-At) / Ac]X 100
where Ac and At are the absorbance of the control and treated samples respectively.
Cell lines and maintenance
Cell lines used in this study MDA-MB-231 (human breast carcinoma), MCF-7 (human breast carcinoma) and WRL-68 (normal human liver embryonic) were attained from National Centre for Cell Science (Pune, India). MDA-MB-231 cells were maintained in L-15 (Leibovitz's) culture medium added with 10 % serum in a humidified atmosphere at 37 ºC without CO2. Whereas, MCF-7 and WRL-68 cells were maintained in Minimum essential medium (MEM) (Eagle) containing Non-essential amino acids at the same condition mentioned above with 5 % CO2. The cell lines were passaged regularly at 70 % confluency.
Assessment of cytotoxicity - XTT assay
Cytotoxicity of the extracts was determined by XTT-formazan dye formation assay (Weislow et al., 1989[52]). MDA-MB-231 and WRL-68 cells (200 µl, 1 x 104 cells/well and 6 x 103 cells/well respectively) were seeded in a 96-well plate with their respective culture medium and incubated at 37 °C for 24 h with/without 5 % CO2 supply. After 24 h, the control wells were replenished with fresh medium and the test wells were added with medium containing 25, 50, 100 and 200 µg/ml of the extract. The plate was re-incubated for 24 h with the same conditions. After incubation period, each well was replenished with 200 µl of fresh medium and 50 µl of XTT (0.6 mg/ml containing 25 µM PMS). The plate was incubated for further 4 h in the same conditions after which the absorbance was measured at 450 nm (with a 630 nm reference filter) in a Dynex Opsys MRTM Microplate Reader (Dynex Technologies, VA, USA). Percentage cytotoxicity was calculated by the following formula:
% Cytotoxicity = [(Ac-At) / Ac] X 100
Ac is the mean absorbance of the control wells and At is the mean absorbance of test wells with a particular extract dosage.
Cellular DNA fragmentation ELISA for apoptosis
Apoptosis was determined by a photometric enzyme-linked immunosorbent assay (ELISA) kit, in which, Bromodeoxyuridine (BrdU) labelled apoptotic DNA fragments in cytosolic extracts of culture supernatants was detected.The experiment was performed as per the supplier's instructions. Cells (MDA-MB-231 and MCF-7) were labeled with 10 µM BrdU at 1 x 105 cells/ml density. 1 x 104 BrdU-labeled cells in 100 µl medium were treated with varying concentrations (12.5, 25, 50, 100 and 200 µg/ml) of the extracts for a period of 4 h. The cells were then lysed and the supernatant containing apoptotic fragments were obtained after centrifugation at 1500 rpm for 10 min and analyzed through ELISA. Obtained sample (100 µl) was transferred to anti-DNA coated 96-well flat-bottom microplate and incubated for 90 min at 15-25 °C. The plate was washed with washing buffer post-incubation and the DNA bound to coated microplate was denatured by microwave irradiation (500 W for 5 min). The wells were then added with 100 µl anti-BrdU-POD (Peroxidase) conjugate and incubated for 90 min.The plate was again washed three times with washing buffer and the wells were added with 100 µl of 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate solution. The plate had been shaken until colour development was sufficient and the absorbance was read at 450 nm after addition of 25 µl stop solution.
CPP32/Caspase-3 activation assay
Caspase-3 activation was determined using CPP32/Caspase-3 Colorimetric Assay Kit (BioVision Incorporated, CA, USA). The assay was performed according to the manufacturer's instructions. MDA-MB-231 and MCF-7 cells (8 x 105 per 12.5 cm2 culture flasks) were seeded with their respective growth medium and incubated for 24 h at 37 °C with/without 5 % CO2. Hydrogen Peroxide (H2O2) (Sigma-Aldrich) was used as a positive control for activation of caspase-3 (Dougherty et al., 2008[17]; Abdelwahab et al., 2011[1]). Confluent cells were added with the respective drug medium (0.1 mM H2O2, 61.6 μg/ml PHR and 76.1 μg/ml PCR) while maintaining an untreated group as negative control, and incubated for 4 h maintaining the same conditions. After treatment incubation, the floating cells were collected from the supernatant. Adherent cells were trypsinized and added to the respective tubes and centrifuged to acquire cell pellet. Tubes were then added with cell lysis buffer and incubated on ice for 10 min. These tubes were again centrifuged at 1000 x g and the supernatant containing cytosolic extracts were transferred to fresh tubes. The protein concentrations of the samples were determined following Bradford assay. 100 μg of each protein sample, diluted to 50 μl with cell lysis buffer, were added in 96 well plates. About 100 μl of reaction buffer, containing 10 mM dithiothreitol (DTT), were added to respective wells and followed by the addition of 5 μl of 4 mM Asp-Glu-Val-Asp (DEVD)- pNA substrate. The plate was incubated at 37 °C for 90 min. Post-incubation the plate was read at 405 nm in a Dynex Opsys MRTM Microplate Reader (Dynex Technologies, VA, USA). Percentage caspase-3 activation was calculated from untreated cells as a control.
% Caspase-3 activation = [(At-Ac) / Ac] X 100
'At' is the mean absorbance of test wells and 'Ac' is the mean absorbance of the control wells with respective extract dosage.
Cell migration inhibition assay
The assay was performed according to the method described by Dimmeler et al. (2000[16]) with some modifications. MDA-MB-231 cells (6 x 105 per well) were cultured on 6-well plates and incubated at 37 °C for 24 h to form a confluent monolayer. In vitro `scratch' wounds were created post-incubation by scrapping the monolayer with sterile cell scrapper. Wells were washed gently with growth medium to remove dislodged cells and were again added with the fresh medium in control wells and medium containing PHR and PCR in treatment wells. The plates were re-incubated at 37 °C further to observe a decrease in distance between the wounded edges at every 4 h intervals (4, 8, 12 and 16 h). Cell migration from the injured monolayer was quantified by measuring the distance between the wound edges before and after injury using a computer-attached inverted phase contrast microscope (Hund Wetzlar, Germany) at five distinct positions (every 5 mm). Percentage cell migration was calculated by the following formula:
% Cell migration = [(D0-Dt) / Dc] X 100
D0 is the mean distance of the scratch wound at time 0 h and Dt is the mean distance of scratch wound after time t (0, 4, 8, 12 and 16 h) with respective extract treatment.
Characterisation and quantification of polyphenol content
Phytochemical screening
Semi-quantitative phytochemical screening was performed to analyse the class of compounds present in the crude extracts. The method employed to screen for different class of chemicals were as described by Trease and Evans (1989[51]), and Ayoola et al. (2008[6]).
Estimation of total phenolic content
Total phenolic content of the extracts were quantified against the standard polyphenol, gallic acid, by the method described by Rajkumar et al. (2010[41]). Amounts of 20, 40, 60, 80 and 100 µg extracts were made up to 0.5 ml with distilled water in separate tubes. An amount of 2.5 ml Folin-Ciocalteau reagent (1:10 dilution) and 2 ml of sodium carbonate (7.5 % w/v) were added to the tubes and incubated at 45 °C for 15 min. Absorbance was then read at 765 nm. Results were expressed in gallic acid equivalence (GAE) in micrograms.
Identification of phenolic compounds through HPLC
Analysis was performed using a Waters 2487 HPLC system consisting of a dual λ detector and a Waters 1525 binary pump, and equipped with a Waters Symmetry® C18 column (5 mm, 4.6 x 150 mm) with Waters SentryTM universal guard column (5 mm, 4.6 x 20 mm) (Waters Corporation, Milford, MA, USA). Phenolic compounds in PHR and PCR extracts of R. emodi were analyzed using the previously reported library of phenolic standards (Sakakibara et al., 2003[46]) as a reference. Gradient elution was performed at 35 °C with solution A (50 mM sodium phosphate (pH 3.3) and 10 % methanol) and solution B (70 % methanol) in the following gradient elution program:
0 - 15 min: 100 % of solution A;
15 - 45 min: 70 % of solution A;
45 - 65 min: 65 % of solution A;
65 - 70 min: 60 % of solution A;
70 - 95 min: 50 % of solution A;
95 - 100 min: 0 % of solution A.
Flow rate was 1 ml/min and injection volume was 20 µl. Detection was monitored at diverse wavelengths (around λmax) for various phenolic compounds, i.e. 250 nm for benzoic acids, isoflavones and most anthraquinones; 280 nm for some flavones, flavanones, catechins, theaflavins and some anthraquinones; 320 nm for cinnamic acids, most flavones and chalcones; 370 nm for flavonols; 510 nm for anthocyanins.
GC-MS analysis for volatile compounds
Instrument specifications
The analysis was performed using Thermo GC - Trace Ultra ver 5.0 coupled with Thermo MS DSQ II, fixed with a DB 5 - MS Capillary Standard column (30 m, i.d.: 0.25 mm, and film thickness: 0.25 μm).
Analytical conditions
Helium was used as a carrier gas (flow-1.0 ml/min). Injector temperature was 260 °C and oven temperature gradually raised from 80 °C to 260 °C (5 °C/min). One μl of sample was injected through injection port and the individual compounds were identified based on their retention times and standard matching spectral peaks available in Wiley mass spectral library.
Statistical analysis
All analyses were carried out in triplicates. For each assay, data were presented as mean ± SD from three independent experiments (n = 3). Statistical analyses were performed by one-way ANOVA and significant differences between groups were determined at P < 0.05. To evaluate relationships between experimental parameters, results were analyzed for correlation and tested for significance by Student's t-test (P < 0.05). MATLAB ver. 7.0 (Natick, MA, USA), GraphPad Prism 5.0 (San Diego, CA, USA) and Microsoft Excel 2007 (Roselle, IL, USA) were used for the statistical and graphical evaluations.
Results
Yield of extracts
Fifty grams of rhizome powder yielded 0.67 g (percentage extract yield: 1.34 % of dry weight) of crude hot petroleum ether extract (PHR) and 0.27 g (percentage extract yield: 0.54 % of dry weight) of crude cold petroleum ether extract (PCR).
Antioxidant potentials of the extracts
Reducing power estimation
The reducing power of a compound depends on the ability of it to donate an electron to break the free radical chain (Duh, 1998[19]). PHR and PCR were able to reduce Fe+3 to Fe+2 as indicated by formation of Perl's Prussian blue. The increase in absorbance corresponds to concentration dependent increase in reducing power of the extracts, PHR and PCR, with butylated hydroxytoluene (BHT) as a standard (Figure 1(Fig. 1)).
DPPH● scavenging activity
Both PHR and PCR exhibited a steady increase in DPPH● scavenging activity, in which the latter was significantly better in DPPH● scavenging. Results were illustrated as % radical scavenging with equivalence to ascorbic acid in micrograms (AAE) (Figure 2(Fig. 2)).
Cell annihilation capability
Cytotoxicity of the extracts was depicted by colorimetric measurement of XTT-formazan dye formation through succinate dehydrogenase of living cells (Weislow et al., 1989[52]). PHR and PCR showcased an increase in % cytotoxicity across the ascending concentration range in both MDA-MB-231 and WRL-68 cells (Figure 3(Fig. 3)).
Apoptosis inductivity
The ability of extracts to induce apoptosis in cancer cells was analysed by quantifying the amount of apoptotic BrdU labeled-DNA fragments released in the cytoplasm of treated cells. Both the extracts entrenched a dose dependent increase in apoptotic fragments in both MDA-MB-231 and MCF-7 cells respectively (Figure 4(Fig. 4)).
Caspase-3 activation
Extracts were investigated to check the involvement of caspase-3 in its apoptosis inductivity. PHR and PCR in their IC50 range (61.6 μg/ml and 76.1 μg/ml respectively) have demonstrated an evident activation of caspase-3 in MDA-MB-231 cells as compared to that of positive control (0.1 mM H2O2) (Figure 5(Fig. 5)). While the drugs H2O2, PHR and PCR did not show significant caspase-3 activation in MCF-7 cells.
Cell migration inhibition
PHR and PCR were tested for its ability to inhibit the migration of metastatic breast cancer cells to prove a characteristic of an anti-metastatic agent. MDA-MB-231 cell migration, in control wells have gradually increased from 0th h to 4th, 8th, 12th and 16th h by protrusion of cells into the open space from edges of scratch wound, to attain a complete confluency at 16th h. Cells with addition of PHR (61.6 μg/ml) and PCR (76.1 μg/ml) displayed minimal migration with some morphological changes till 16th h, after which no further migration was observed (Figure 6(A)(Fig. 6)). Percentage cell migration was portrayed in Figure 6(B)(Fig. 6) showing significant (P < 0.05) difference between the control and treated groups in its migration.
Quantification and characterisation of polyphenol content
Phytochemical screening
Screening of the extracts revealed the presence of various groups of chemical compounds. The hot extracts were found to possess additional number of chemical groups when compared to cold extracts (Table 1(Tab. 1)).
Total phenolic content of the extracts
Results were signified in GAE (in micrograms) as depicted in Table 2(Tab. 2). PHR was noted to harbor high concentration of phenolics when compared to PCR. The total phenolic contents were positively correlated (at P < 0.05) to that of the antioxidant efficacies of the extracts conforming that the soundness of antioxidant activity was due to the presence of polyphenol in the extracts (Table 3(Tab. 3)).
HPLC analysis
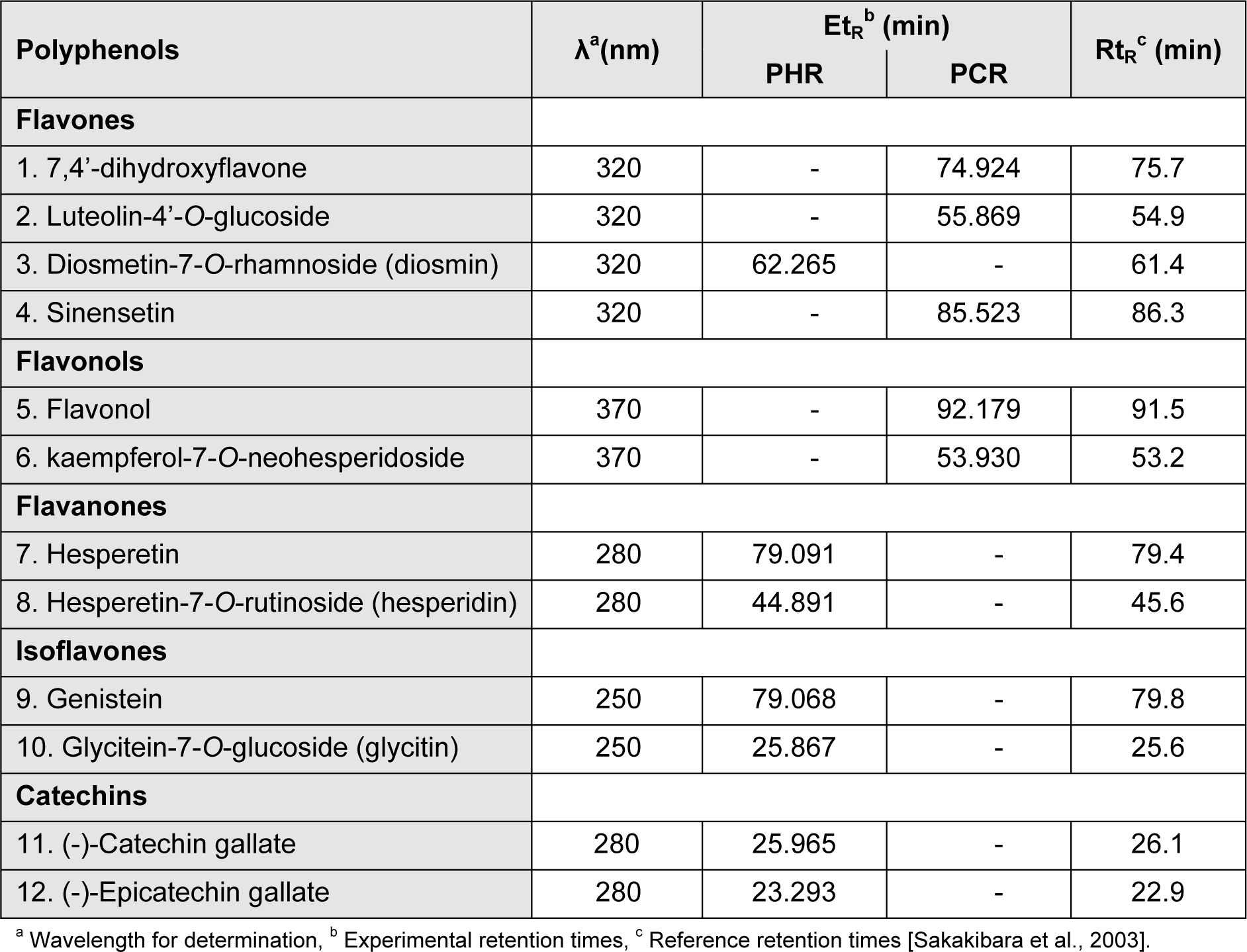
It is tricky to characterize each and every compound in the crude extracts due to diversity and complexity of natural phenolic compounds (Surveswaran et al., 2007[50]). Accordingly, varied polyphenolic classes were identified in PHR and PCR, using the reported library of polyphenolic standards with analytical characteristics such as λmax, retention time, determining λ, slope and limit calibration (Sakakibara et al., 2003[46]). Phenolics identified were portrayed in the Table 4(Tab. 4). Both the extracts were also found to contain few unknown compounds as apparent from the HPLC chromatograms, whose isolation and identification is in prospect.
GC-MS analysis
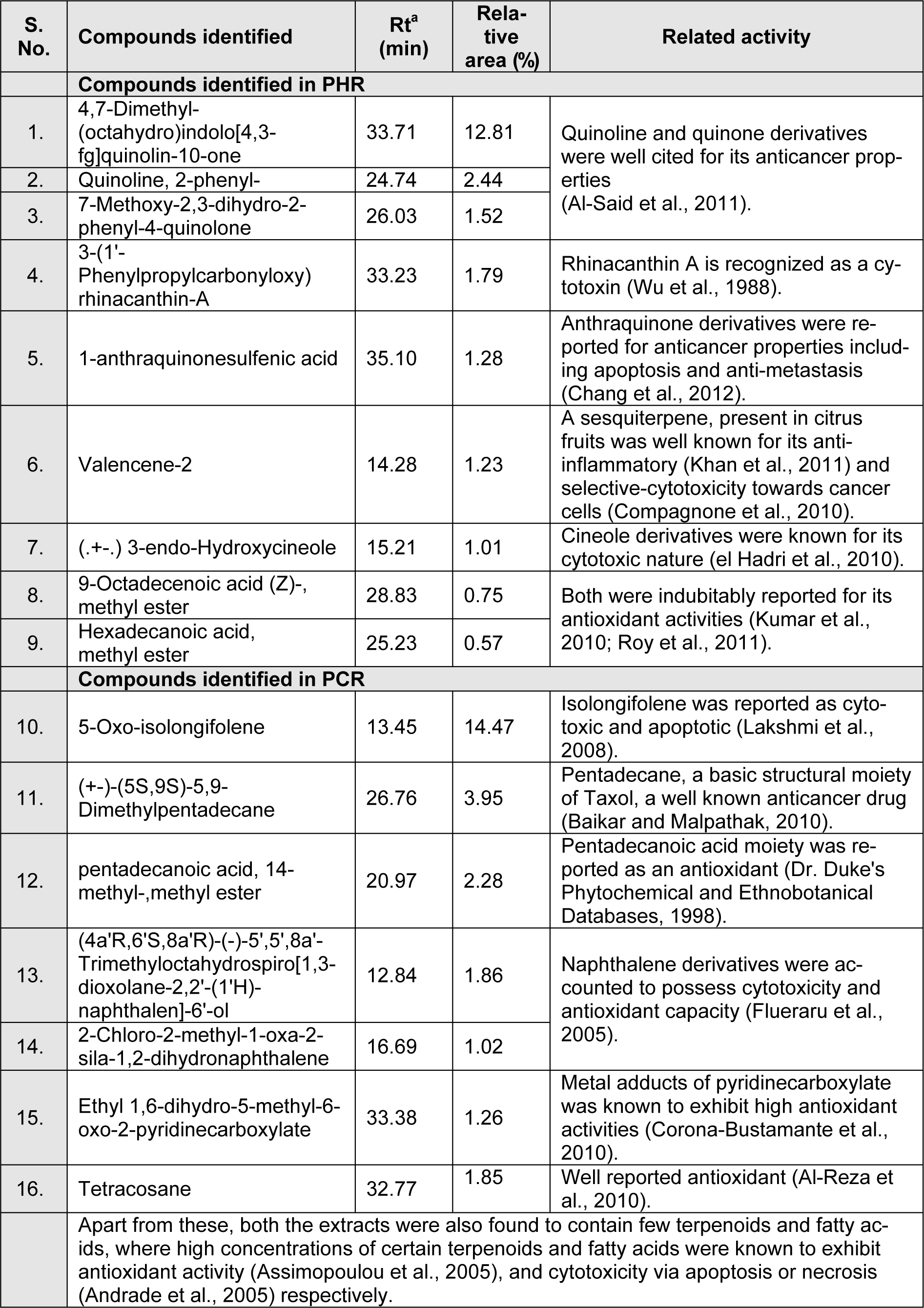
The GC-MS analysis of the samples PHR and PCR had revealed the presence of various volatile components. The major components along with their previously reported bioactivities were given in Table 5(Tab. 5).
Discussion
Plants were being used as a source of chemopreventives since decades. Many plant products have been proved to be effective against carcinogenesis in the forms of dietary supplements, herbal formulations, concoctions or pure phytochemicals which act synergistically to give higher efficiency with lesser side effects (Desai et al., 2008[15]). R. emodi was being used as a food source and also to meet medical complications, which later was given with scientific evidence to substantiate the preliminary activities (Babu et al., 2003[7]; Rajkumar et al., 2010[41]; 2011[42]; Kounsar et al., 2011[30]). However, these studies have reported activities without attempting to define the mechanism of action or chemically characterising the derivation of bioactivity. Our study reports specific anticancer activities of R. emodi by defining a possible mode of action along with its chemical components identified through HPLC and GC-MS techniques. Petroleum ether extracts of R. emodi, both in their hot and cold counterparts exhibited significant (P < 0.05) therapeutic properties in all the test systems analysed.
PHR and PCR have shown markable antioxidant activities through DPPH and reducing power test systems. The antioxidant property of any agent depends upon its ability to quench the free radicals generated in the test system through reduction reaction (Duh, 1998[19]). Accordingly, PCR had exhibited significantly better free radical scavenging when compared to PHR in both the antioxidant assays (Figure 1(Fig. 1) and Figure 2(Fig. 2)). The cytotoxicity of the extracts had imparted a contrasting result, where PHR was more potent than PCR in its cell annihilation capacity. The 50 and 100 µg/ml doses of PCR and 100 µg/ml of PHR have exhibited significantly (P < 0.05) higher cytotoxicity in cancer cells (MDA-MB-231) when compared to non-tumoral cells (WRL-68) (Figure 3(Fig. 3)), the property which can be used to specifically target cells by imparting lesser side effects to normal cells in the body.
The drugs with the ability to exterminate cancer cells are efficient in chemotherapy of cancer (Mooney, 2005[35]) but still to study the mechanism behind the toxicity, these extracts were tested for its ability to induce apoptosis. Apoptotic induction is considered as a specific therapeutic approach towards cancer chemotherapy, since cancer cells lack apoptosis due to dysfunction of the cell cycle regulation along with either an overproliferation of cells and/or decreased removal of cells (Elmore, 2007[21]). PHR and PCR have shown evident apoptosis induction in both the cell lines tested (MDA-MB-231 and MCF-7) (Figure 4(Fig. 4)). Apoptosis induction ability of the extracts was not studied in WRL-68 cells, as these cells were used in cytotoxicity testing; only to prove the cancer-cell-specificity of the extracts. However, since the study deals with anti-breast cancer potential of the extracts, MCF-7 cells were included in rest of the studies. These extracts were found to be efficient against MDA-MB-231 cells in comparison with MCF-7, where the latter appeared to be more resistant to apoptosis (Calcabrini et al., 2006[10]; Zhong et al., 2009[56]). The exact mechanism behind the activity is not known, yet the extracts were consolingly specific to induce apoptosis in estrogen receptor (ER)-negative cell system when compared to its ER-positive counterpart.
The extracts were tested for its ability to activate caspase-3 in MDA-MB-231 and MCF-7 cells. Both PHR and PCR have resulted to significantly activate caspase-3 in MDA-MB-231 cells (Figure 5(Fig. 5)) but not in MCF-7 cells.As caspase-3 was naturally absent in wild MCF-7 cells due to a deletion in the 47 base-pair region in CASP-3 gene (Janicke, 2009[27]), the extracts would have induced apoptosis in MDA-MB-231 through caspase-3 activation. This result was found concomitant to that of previous report (Mandlekar et al., 2000[34]).
The extracts were also tested to inhibit cancer cell migration, which is an attribute of metastasis. Consequently, both extracts inhibited the migration of metastatic breast cancer cells (MDA-MB-231; Figure 6A(Fig. 6)). Unlike control group, the PHR and PCR treated cells exhibited decreased % cell migration at regular time intervals observed (4, 8, 12 and 16 h; Figure 6B(Fig. 6)), and unveiled some morphological changes with complete arrest of cell migration at 16th h. Similar results were reported by Yang et al. (2011[54]) and Naveen Kumar et al. (2012[36]). In our study, PHR was efficient compared to PCR, in inhibiting cell migration at every interval. This property of the extracts concedes that PHR and PCR could be considered as a source to derive active therapeutic components to be used in treating advanced malignancies of breast cancer.
Both the extracts were further characterised to reveal the phytochemical constituents. For that reason, the extracts were screened semi-quantitatively which divulged the presence of various chemical groups. The assessed total phenolic content of the extracts have also showed a strong positive correlation with antioxidant potentials of the extracts. Similar correlations have been previously reported by Guha et al. (2010[25]). Consequent to this, the extracts were analysed through HPLC to uncover the major polyphenols present. PHR and PCR were found to contain 12 polyphenols collectively (Table 4(Tab. 4)). The high toxicity of PHR is thus justified by the presence of catechins (Guha et al., 2010[25]) and the high antioxidant activity of PCR is defensible by flavones and flavonols present in it (Ramamoorthy and Bono, 2007[43]). Except flavonol, other compounds are reported for the first time in R. emodi.
Furthermore, the GC-MS analysis of PHR and PCR has revealed the presence of diverse volatile components holding high therapeutic values, hitherto unreported in R. emodi. PHR was found to majorly contain
- 4,7-Dimethyl-(octahydro)indolo[4,3-fg]quinolin-10-one (12.81 %) followed by
- Quinoline, 2-phenyl- (2.44 %), 7-Methoxy-2,3-dihydro-2-phenyl-4-quinolone (1.52 %),
- 3-(1'-Phenylpropylcarbonyloxy) rhinacanthin-A (1.79 %),
- 1-Anthraquinonesulfenic acid (1.28 %), Valencene-2 (1.23 %),
- (.+-.) 3-endo-Hydroxycineole (1.01 %),
- 9-Octadecenoic acid (Z)-, methyl ester (0.75 %) and
- Hexadecanoic acid, methyl ester (0.57 %),
whereas, PCR was found to be rich in
- 5-Oxo-isolongifolene (14.47 %),
- (+-)-(5S,9S)-5,9-Dimethylpentadecane (3.95 %),
- Pentadecanoic acid, 14-methyl-, methyl ester (2.28 %),
- (4a'R,6'S,8a'R)-(-)-5',5',8a'-Trimethyloctahydrospiro[1,3-dioxolane-2,2'-(1'H)-naphthalen]-6'-ol (1.86 %),
- 2-Chloro-2-methyl-1-oxa-2-sila-1,2-dihydronaphthalene (1.02 %),
- Ethyl 1,6-dihydro-5-methyl-6-oxo-2-pyridinecarboxylate (1.26 %) and
- Tetracosane (1.85 %).
These compounds might have played a role in diverse anticancer activities showcased by PHR and PCR, where such justifications were also made by Patil et al. (2009[38]). As mentioned in Table 5(Tab. 5), the compounds identified through GC-MS, directly or its derivatives/analogues hold therapeutic potentials related to the activities of extracts explicated in this study.
These results, together suggests that the R. emodi extracts exhibits cancer-cell-specific cytotoxicity in ER-negative breast cancer cells, through apoptotic induction mediated by caspase-3 activation along with evident cell migration inhibition potential, and the active principles which have been identified by HPLC and GC-MS analyses were possibly responsible for their mode of action.
Resultantly, R. emodi petroleum ether extracts accommodate various biological properties which exemplify significant antioxidant property, cancer-specific cytotoxicity, ER-negative cell selective-apoptosis inductivity, potent caspase-3 activation and evident cell migration inhibition potential. Further, the extracts were also found to be rich in polyphenols, terpenes, and fatty acids thus endorsing the reason for its diverse therapeutic potentials. These extracts could persuasively serve as a source harbouring specific therapeutic compounds to treat breast cancer in its higher malignancy, and thus necessitates its use in dietary-based anti-breast cancer therapies.
Acknowledgements
We are thankful to the management of VIT University, Vellore, Tamil Nadu, India, for funding this research work and for providing the necessary infrastructure for the successful completion of the same.
References
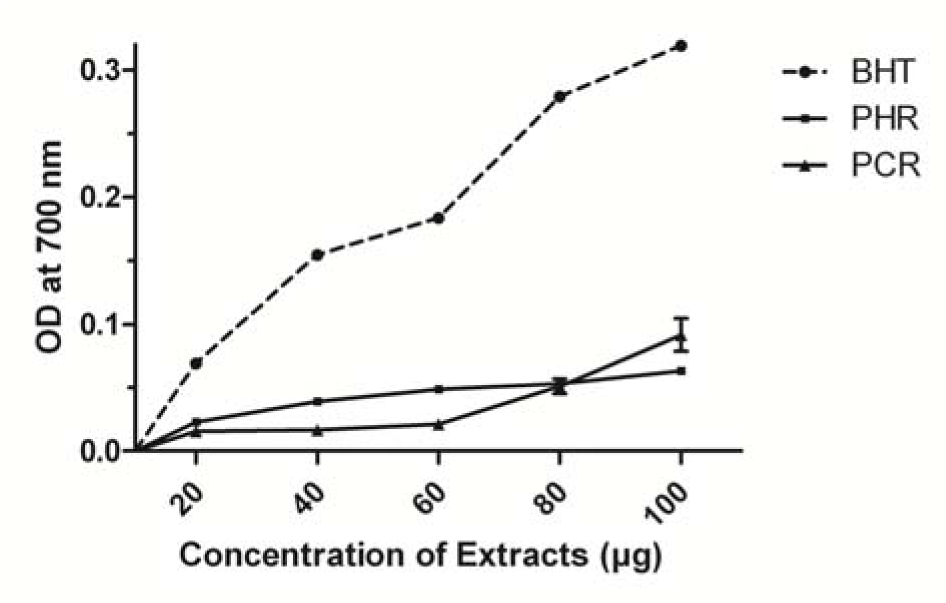
Figure 1: Reducing power of PHR and PCR with BHT as standard. Data points are mean values ± SD (n = 3, P < 0.05).
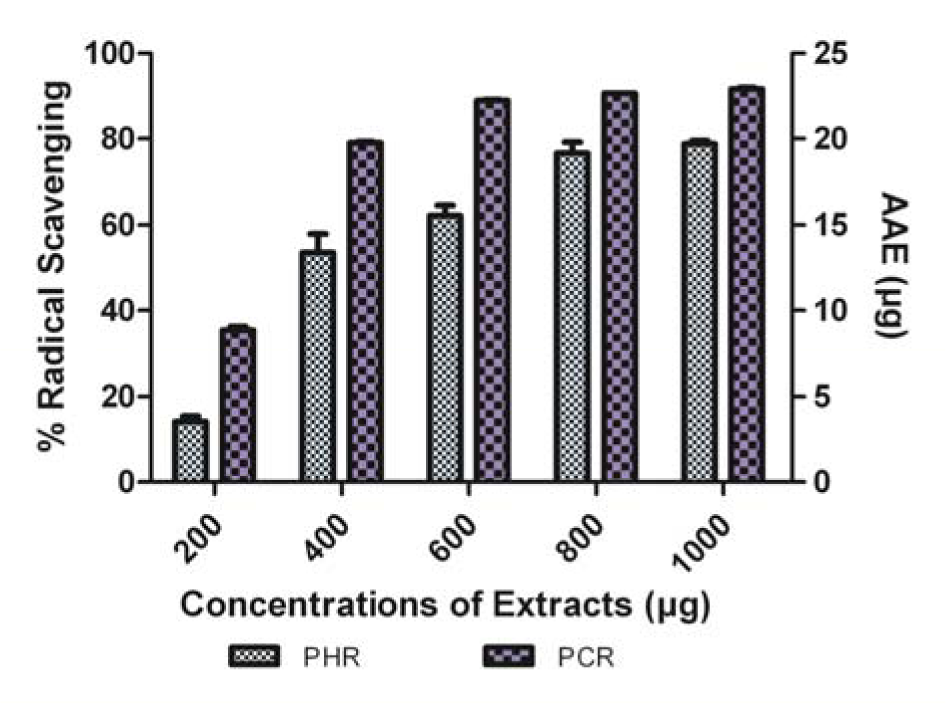
Figure 2: Comparison of DPPH● scavenging ability of PHR and PCR expressed in AAE. Data expressed as % radical scavenging (mean ± SD, n = 3, P < 0.05).
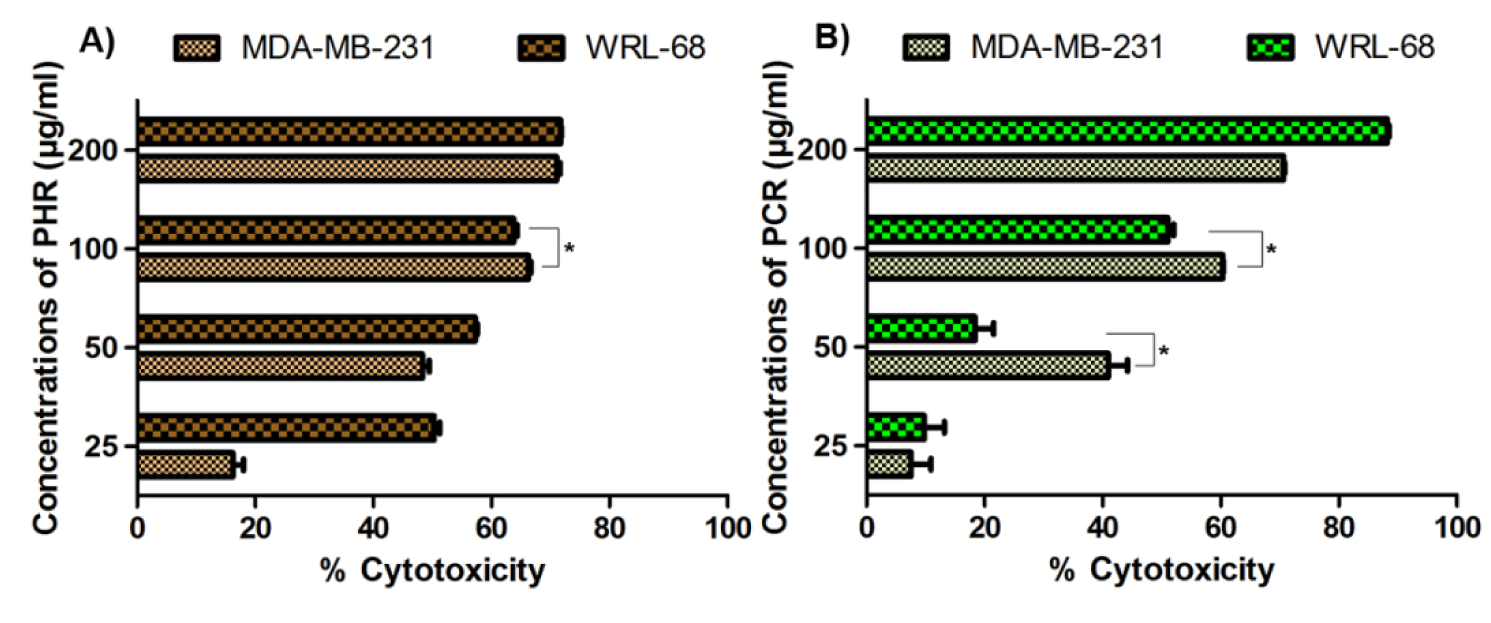
Figure 3: % Cytotoxicity of PHR and PCR on MDA-MB-231 and WRL-68 cell lines expressed as mean ± SD (n = 3). Asterisk (*) indicates a significantly (P < 0.05) lesser cytotoxicity in WRL-68 cells compared to MDA-MB-231 cells.
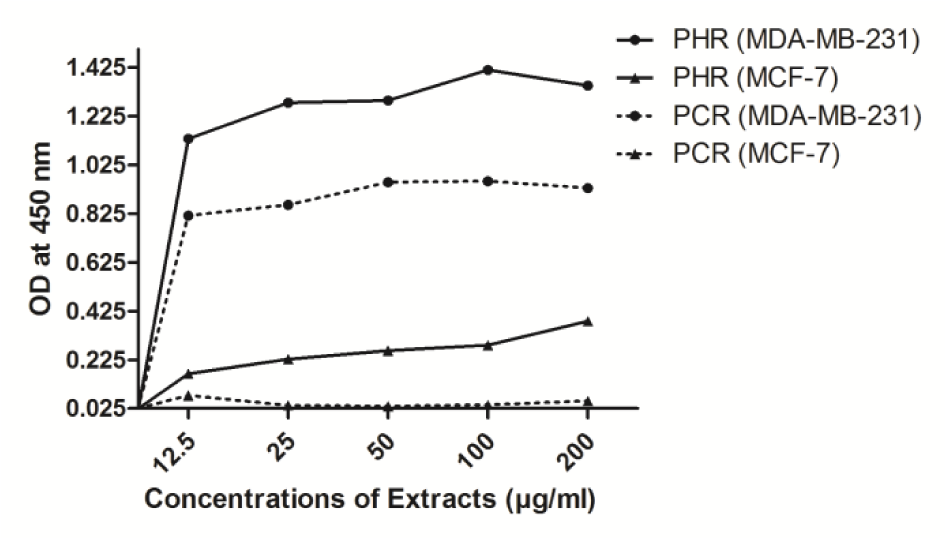
Figure 4: Induction of apoptosis corresponding to measure of apoptotic DNA fragments in MDA-MB-231 and MCF-7 cells by PHR and PCR treatment, expressed as OD ± SD (n = 3).
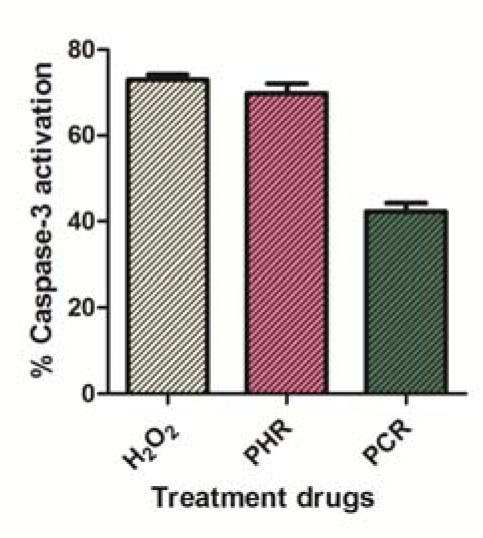
Figure 5: % Caspase-3 activation in MDA-MB-231 cells by 0.1 mM H2O2, 61.6 μg/ml PHR and 76.1 μg/ml PCR. Data expressed as mean ± SD (n = 3).
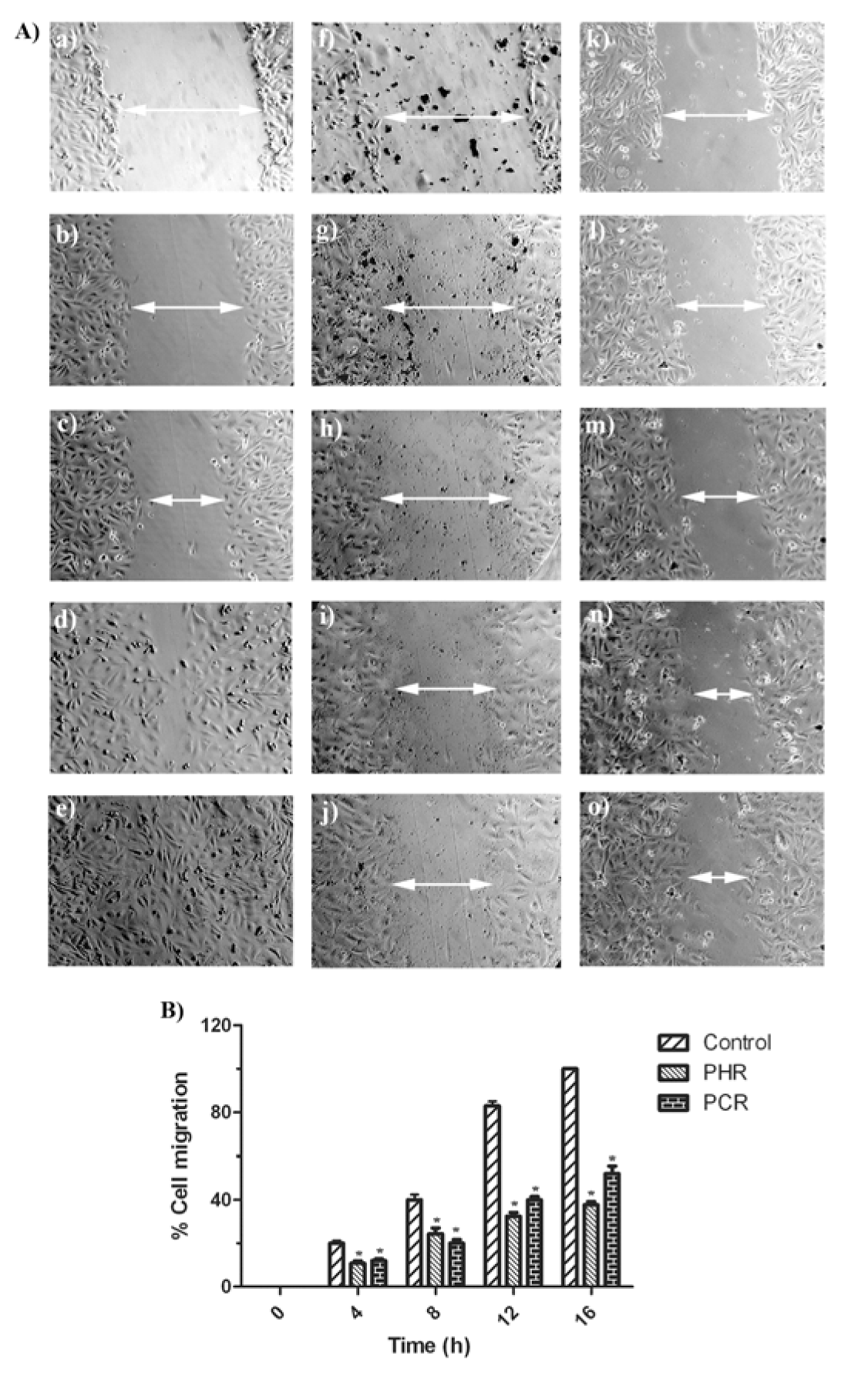
Figure 6: (A) Observation of MDA-MB-231 cells migration, from edges of the scratch wound after 0, 4, 8, 12 and 16 h in control well (a, b, c, d, e), PHR treated well (f, g, h, i, j) and PCR treated well (k, l, m, n, o) respectively (n = 3). (B) Calculated % cell migration of MDA-MB-231 cells, portraying decreased migration in PHR and PCR treated cells compared to control cells. Data presented as mean ± SD (n = 3). Asterisk (*) indicates significant (P < 0.05) difference from the respective control groups.
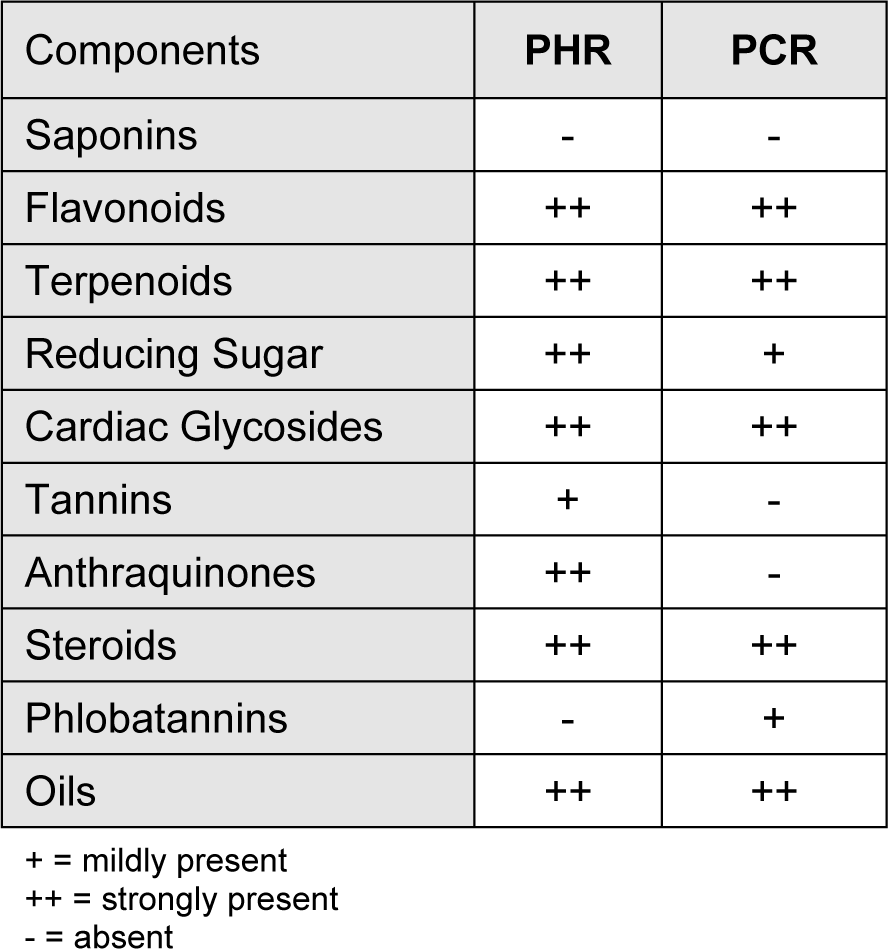
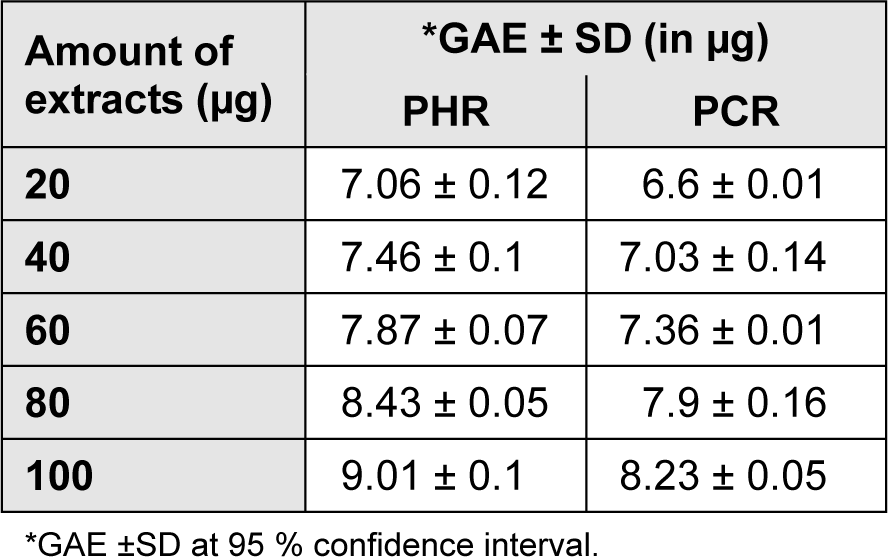
Table 2: Total phenolic content of R. emodi extracts given in mean ± SD (n = 3, P < 0.05). GAE of the extracts is expressed in micrograms.
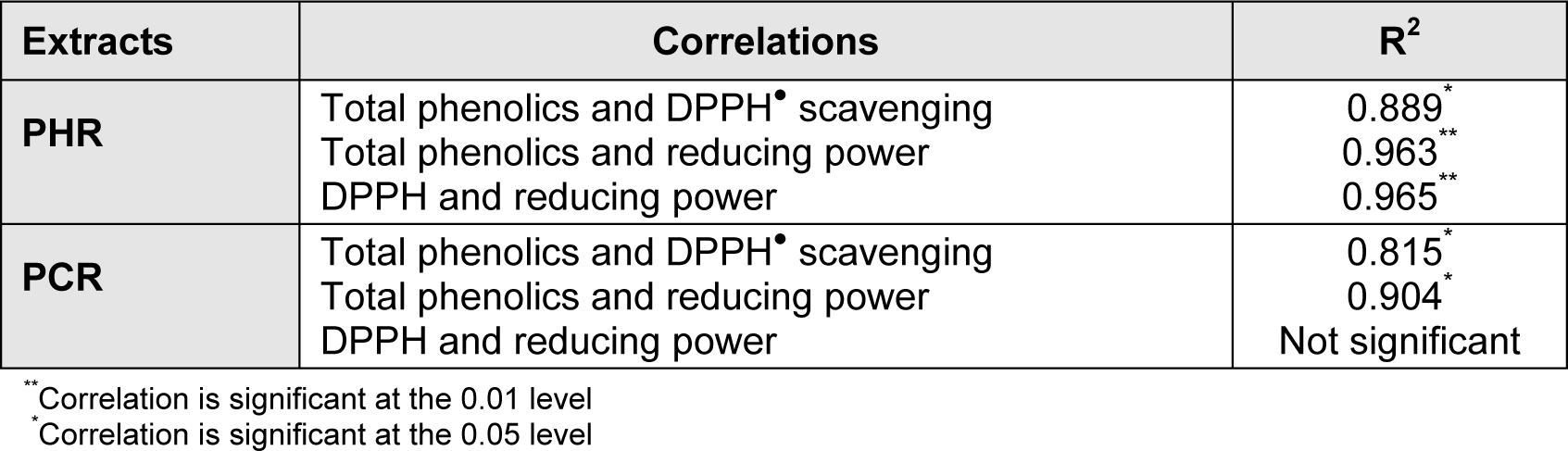
Table 3: Correlations between experimental results (of total phenolic estimation, reducing power, DPPH) tested for significance. R2 denotes coefficient of determination.
[*] Corresponding Author:
R. Ashok Kumar, Department of Zoology, Government Arts College, Dharmapuri - 636 705, India; Tel: +91 9994333688, eMail: ashoku_2000@yahoo.com