Review article
Three dimensional electron microscopy and in silico tools for macromolecular structure determination
Subhomoi Borkotoky1, Chetan Kumar Meena1, Mohammad Wahab Khan1, Ayaluru Murali1[*]
1Centre for Bioinformatics, School of Life Sciences, Pondicherry University, Puducherry-605014, IndiaEXCLI J 2013;12:Doc335
Abstract
Recently, structural biology witnessed a major tool - electron microscopy - in solving the structures of macromolecules in addition to the conventional techniques, X-ray crystallography and nuclear magnetic resonance (NMR). Three dimensional transmission electron microscopy (3DTEM) is one of the most sophisticated techniques for structure determination of molecular machines. Known to give the 3-dimensional structures in its native form with literally no upper limit on size of the macromolecule, this tool does not need the crystallization of the protein. Combining the 3DTEM data with in silico tools, one can have better refined structure of a desired complex. In this review we are discussing about the recent advancements in three dimensional electron microscopy and tools associated with it.
Keywords: cryo-EM, single particle analysis, CTF correction, segmentation
Introduction
Recent past witnessed the emergence of new tool in structural biology in the form of electron microscopy and single particle analysis. It complemented the currently existing tools namely, X-ray crystallography and nuclear magnetic resonance (NMR). This tool has several advantages over the conventional structural biology tools (X-ray crystallography and NMR) such as, structure elucidation without crystallization, solving the structure in physiological conditions, and with literally no upper limit on size of the protein. Though greater purity is of high demand, this technique needs low concentrations of the protein (typically of the order of 1 ng/μl for a protein of ~100 kDa molecular weight for negative stain). However, despite of proving its ability in all corners of structural biology, three dimensional transmission electron microscopy (3DTEM) has relatively fewer entries in RCSB compared to deposits from crystallography and NMR. The present review article was taken up to give an outline of the single particle analysis with few examples where 3DTEM was found to be unique. Also this review article presents a brief account on other promising online and offline tools that will complement the 3DTEM technique.
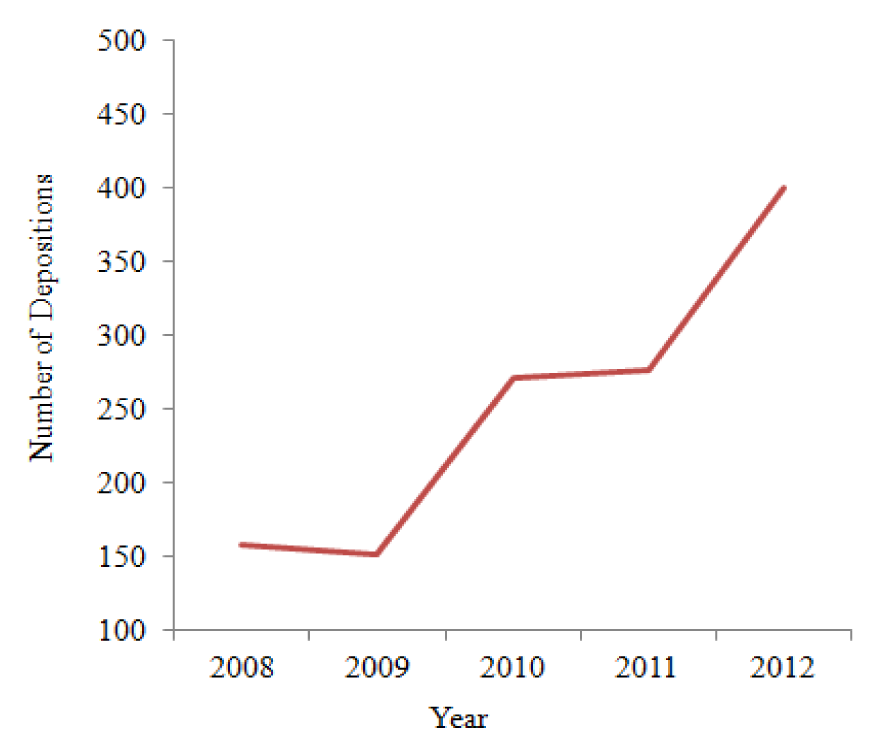
As per advancements of more sophisticated instruments and in silico tools over recent years, electron microscopy is getting a greater push to the arena of solving molecular structures, along with other methods like X-ray crystallography and nuclear magnetic resonance (NMR). As compared to the prominent structure exploration methods like X-ray crystallography and NMR, 3DTEM techniques are the structural biologists' best choice for larger macromolecular assemblies (having molecular weights greater than 60 kDa) and other larger cellular components. There is no need for growing 2D crystals for 3DTEM methods; it mainly relies on the symmetry properties of particle (Jonic et al., 2008[33]). The increase in the number of electron microscopy (EM) solved structures is evident from the collaboration of the ftp archives of Protein Data Bank (PDB) and EM Data Bank (EMDB) to facilitate user access to EM maps and models (Berman et al., 2013[9]). According to EM databank statistics, 1749 entries were deposited in its database (http://www.emdatabank.org/recententries.html) till February, 2013 and with a gradual increase in data submission in EMDB in the last five years (Figure 1(Fig. 1)).
Since the electron microscopy technique is a classic technique and known to scientists in all branches of life sciences, a huge collection of literature exists and it is not possible to include all of it. We focus on single particle analysis (SPA) - a tool that works with electron microscopy, which has laid its foundation in 1970s and started gaining the attention of more researchers since 1995 (Frank, 2009[21]). In view of the space restriction, the present review considers only the developments in the recent years. Readers might refer to the earlier reviews that appeared on similar topics for complete coverage of the literature on this subject (Jiang and Ludtke, 2005[32]; Llorca, 2005[39]; Ruprecht and Nield, 2001[55]).
The present review mainly focuses on the specimen preparation approaches for electron microscopy with an introduction of single particle analysis, some important tools that are available to complement the 3DTEM and concludes with some notable reports to prove the uniqueness of 3DTEM.
Specimen preparation methods for electron microscopy
The most commonly used staining methods in electron microscopy are negative staining, CryoEM and Cryo-negative staining. Each of these three methods plays a considerable role in resolution of the data and contrast of the images. A detailed account of these methods is listed below.
Negative staining
Among all the staining methods available, negative staining (NS) is the simplest one. First reported in 1959 (Brenner and Horne[13]), it was revisited by several researchers. Generally, the aqueous solutions of heavy metal salts are used as stains which make the sample to appear darker than the background, so the term “negative staining” is attributed to this process. Most commonly used negative stain is uranyl acetate which gives high contrast. The other compounds that are in use are sodium/potassium phosphotungstate, uranyl formate, ammonium molybdate (Bremer et al., 1992[12]), as well as patented stains, such as NanoVan® and NanoW® (Hainfeld et al., 1994[26]). Negative staining can reveal the true solvent-excluded surface and shape of a biological molecule. Proper identification of intra-molecular information by negative staining, such as alpha-helices and/or beta-sheets are quite theoretical, which relies upon the relatively large mass-thickness difference between the biological material and the surrounding stain (De Carlo and Harris, 2011[17]). The high vacuum that is required to be maintained in a typical TEM column can deform the biological samples by collapsing/flattening effect which is mainly due to the vacuum drying.
Cryo-freezing
It is well known that the negative staining approach always results in a low resolution data (typically of the order of 1.5 - 2 nm). CryoEM (also known as Cryo-freezing) is an alternative where the nativity of the macromolecule is preserved and considerably high resolution is achieved. Also, Cryo-freezing eliminates many of the artifacts that may disturb the structural integrity of the biological macromolecules. It allows vitrification of biological samples in their native state and the freezing process is done so rapidly that no ice crystals will be formed during the sample preparation (Dubochet et al., 1982[18]). There are different methods of preparing frozen, hydrated biological specimens such as - sandwich between folding grids, sandwich between two carbon films, sandwich between pretreated behenic acid film and poly lysine coated film, sandwich between grids of different meshes (Jaffe and Glaeser, 1984[29]; Talmon et al., 1979[64]; Taylor and Glaeser, 1973[65]) in which manual freezing is done at pretreated carbon films in liquid nitrogen. Other methods like thin film freezing (Adrian et al., 1984[3]) use pretreated carbon films or holey grids for support and liquid ethane as coolant. This approach is known to give higher resolution structural details but suffers from poor contrast and high radiation damage. The poor contrast that is inherent in the cryoEM sets a lower limit (of about 200 kDa) on the protein size.
However, there are some attempts to work-around by generating focal pairs (Ludtke and Chiu, 2003[42]). In this approach, two micrographs were taken for the same image - one with true focus (poor or no contrast) and one with greater defocus (having better contrast with almost loss of resolution of the image). The images from the true focus, as identified by transferring the coordinates of the images from micrographs of greater defocus, will be used for analysis.
Cryo-negative staining
Cryo-negative staining combines the advantages of both negative staining and cryo-EM. It uses stains like ammonium molybdate just before vitrification (Adrian et al., 1998[2]). It has many advantages over traditional techniques such as better signal to noise ratio, better stability of the specimen, rapid collection and analysis of images, reduced radiation damage and lower range of sample size. Recent studies on Limulus SAP-like pentraxin (Shrive et al., 2009[59]) and human Pol II complex (Kassube et al., 2012[34]) uses the cryo-negative staining approach and reported structures to the tune of 14 Å and 25 Å resolution respectively. There are however, some limitations of cryo-negative staining. Few biological assemblies may show sensitivity to the saturated stain, or it may overemphasize the contrast of low-resolution features (De Carlo et al., 2002[16]; Jawhari et al., 2006[30]).
Single particle analysis
Single particle analysis (SPA) is one of the novel approaches for reconstructing theoretical models of large proteins and became popular in recent years. The term “single particles” refers to the unique views of the projection images that are isolated and unordered (Frank, 2002[20]). The main aim of single particle reconstruction is to generate a 3D model from 2D images. The combination of SPA and EM is capable of revealing information related to conformational changes between various states (Rouiller et al., 2002[54]), morphological characterization (Ludwig et al., 2003[43]), DNA break repair (Spagnolo et al., 2006[62]) etc. There is also evidence for combining nanotechnology and SPA to study important biological problems like self-assembly of virus-like particles (Sun et al., 2007[63]), and the influence of charge and size variables on the templated assembly of virus capsids (Daniel et al., 2010[15]).
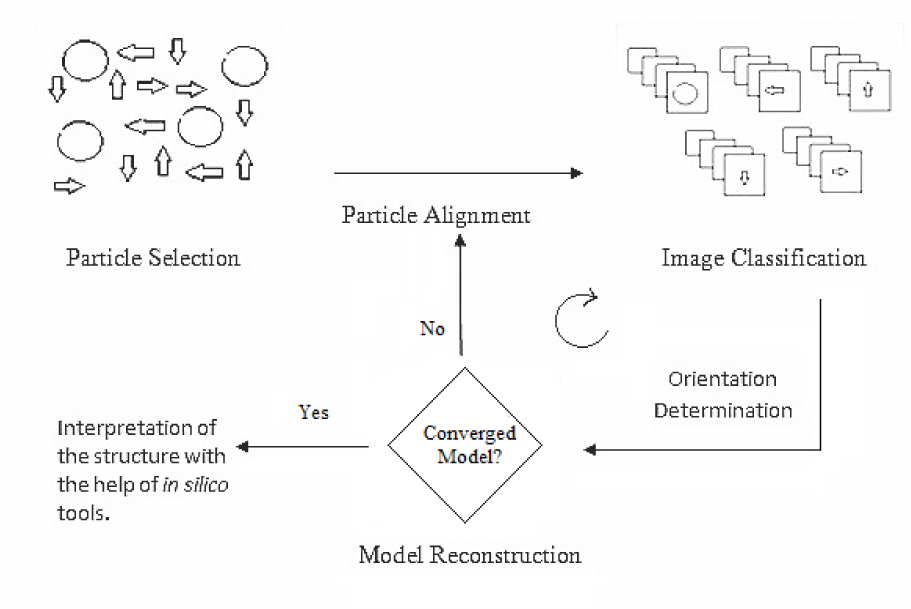
The single particle reconstruction is an iterative process that can be divided into several common steps which include selection of individual projections of the particles from the electron micrograph, classification of particles with identical views and their alignment, orientation determination by comparing each particle to a set of reference projections, and finally reconstruction of an initial three dimensional model. The model thus generated in the first iteration (initial model or density map), can be used as a reference for the reconstruction process by repeating the above mentioned steps, until one gets a useful stable final model. Occasionally, one would also like to apply contrast transfer function (CTF) correction (see next section) in order to improve the resolution of the model. With the final model in hand, one can use various in silico tools for visualization and structure interpretation (Thuman-Commike, 2001[66]). A graphical workflow of this process is given in Figure 2(Fig. 2).
In silico tools for 3DTEM data analysis
In this section we will discuss some of the in silico tools and packages available for 3DTEM data analysis.
Tools for generating the electron density maps
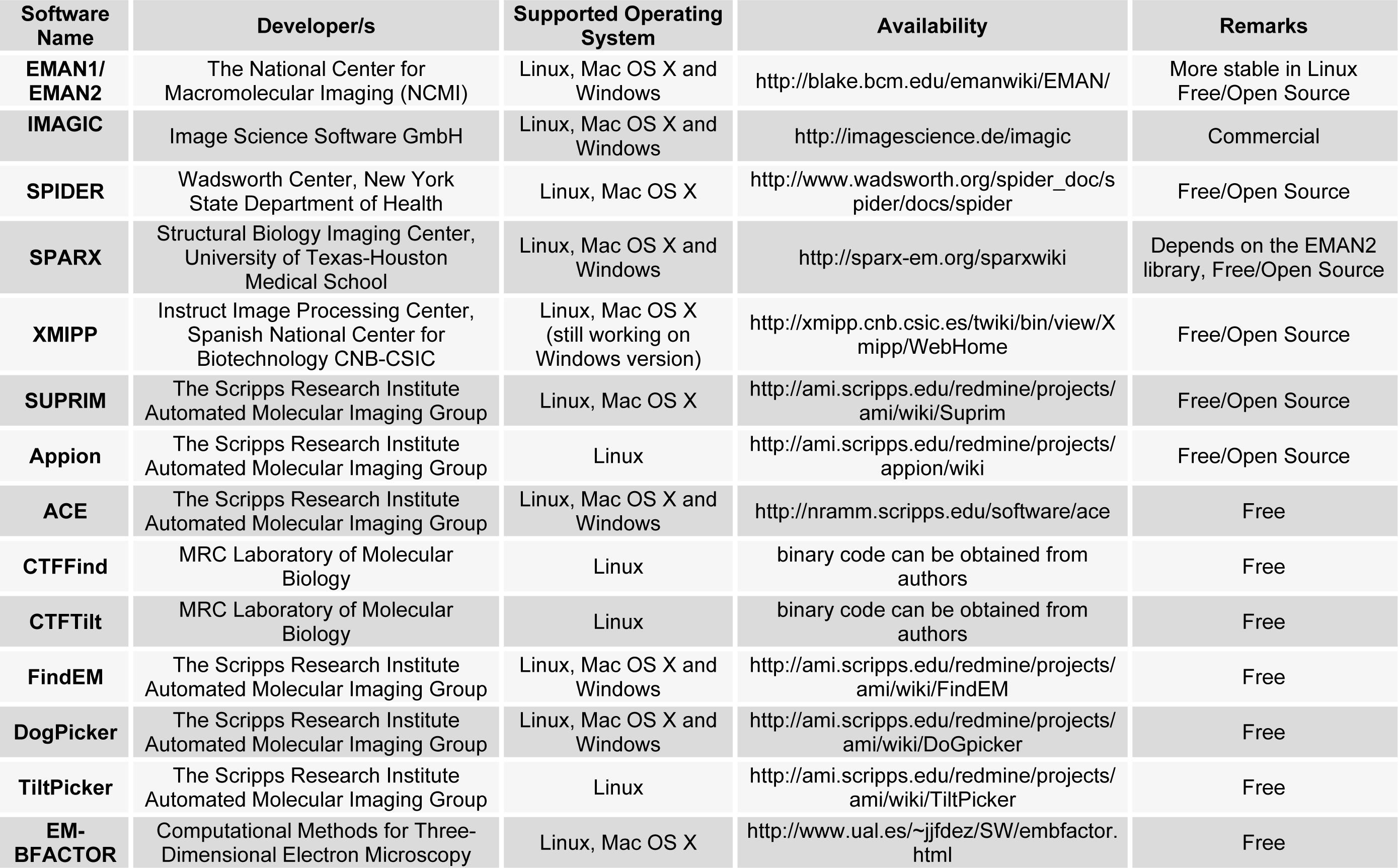
Software packages available for single particle analysis are mainly EMAN1/ EMAN2 (Ludtke et al., 1999[41]), IMAGIC (van Heel et al., 1996[70]), SPIDER (Frank et al., 1996[22]), SPARX (Hohn et al., 2007[27]), XMIPP an X-Window-based Microscopy Image Processing Package (Sorzano et al., 2004[60]), SUPRIM (Schroeter and Bretaudiere, 1996[58]).
Apart from these, one can use the pipeline, APPION (Lander et al., 2009[37]), which includes the packages ranging from single particle analysis, tomography to image analysis tools. Few of them are discussed below:
- Single particle analysis [EMAN, SPIDER, FREALIGN (Grigorieff, 2007[25]), IMAGIC and XMIPP], and
- Tomography [IMOD (Kremer et al., 1996[35]) and ProTomo (Winkler, 2007[76])].
- Particle picker: In general, one has to pick-up several thousands of well-isolated particles from electron micrographs for reconstruction purpose. Few automated tools such as FindEM (Roseman, 2004[51]), DoGPicker and TiltPicker (Voss et al., 2009[74]) are available which will be useful when handling a huge number of electron micrographs.
- CTF correction: Contrast transfer function (Yi et al., 2004[78]), also known as CTF correction is an important step in the SPA that one has to pay attention to, in order to get a high resolution model. This becomes very important especially when one deals with CryoEM data. Tools such as ACE (Mallick et al., 2005[44]), CTFFind and CTFTilt (Mindell and Grigorieff, 2003[45]) are available for arriving at the right CTF.
- Resolution measurement: The most common practice for measurement of resolution of the refined model is 0.5 cut-off in the Fourier Shell correlation (FSC), meaning to take the point at which the FSC drops below 0.5 (Booth et al., 2004[11]). Almost all major SPA packages listed above, including EMAN, provide inbuilt tools to generate these FSCs. Apart from these, Sousa and Grigorieff (2007[61]) designed a novel tool - RMEASURE - to estimate the resolution of the refined maps by calculating the correlation between neighbouring pixels of the model in Fourier space.
- Bfactor correction: Bfactor, also known as temperature factor is another important parameter in context to protein structure as it indicates relative vibrational motion of different parts of the structure. EM-BFACTOR (Fernandez et al., 2008[19]; Rosenthal and Henderson, 2003[52]) is one of the tools that allows automatic determination of the Bfactor for maps at a resolution higher than 10 Å, and allows to sharpen the map and compensate the decay of the amplitudes.
A brief summary of these tools is given in Table 1(Tab. 1).
Software/packages for working with the electron density maps generated by SPA
Having the model of a macromolecule (or macromolecular complex) successfully generated using SPA, one would like to work around the model in several aspects. For visualization of electron density maps one can use Chimera (Pettersen et al., 2004[46]), Amira (Pruggnaller et al., 2008[49]), Avizo (Westenberger, 2008[75]), IMOD (Kremer et al., 1996[35]) and VMD (Humphrey et al., 1996[28]). Among these, Amira and Avizo are commercial packages.
At times one may have to deal with the density map of a macromolecule in its oligomeric form. In such case, segmentation of individual monomers is of utmost importance. Manual segmentation of density maps can be done with Amira, Avizo and Chimera, while automatic segmentation can be done with EMAN, CoDiv (Volkmann, 2002[73]), VolRover (Baker et al., 2006[6]) and Segger (Pintilie et al., 2010[47]) plug-in implemented with Chimera. Segmentation procedure is mainly done for extracting a particular region or density of interest from a map of macromolecular assembly. In most of the tools (e.g., Chimera, CoDiv), the segmentation of maps is done by watershed transform (Beucher and Lantuejoul, 1979[10]; Vincent and Soille, 1991[72]). One can validate the individual subunits identified from automated segmentation by fitting them with their corresponding high-resolution crystal structures and/or manually segmented subunits using foldhunter (Jiang et al., 2001[31]).
When both the Cryo-EM map of a macromolecule and its homology model or high resolution crystal structure are available, one can use fitting studies for getting a more detailed view of structural characterization of a molecular assembly. MODELLER, which is a homology modelling tool commonly used with X-ray and NMR structures as templates, also incorporates two fitting modules - Mod-EM for rigid fitting (Topf et al., 2005[68]) and Flex-EM for flexible fitting (Topf et al., 2008[69]). Similarly, one can also use the SITUS package for fitting crystal structures by both rigid body and flexible way (Wriggers et al., 1999[77]). Other tools that can be used for docking atomic models into EM maps are Dock-EM (Roseman, 2000[50]), EMFit (Rossmann et al., 2001[53]), Foldhunter (Jiang et al., 2001[31]), Fit in Map and MultiFit module in UCSF Chimera (Goddard et al., 2007[24]; Tjioe et al., 2011[67]), ADP_EM (Garzon et al., 2007[23]) etc. Segger plug-in for the UCSF Chimera also facilitates rigid-body docking of models into density maps using segmented regions and it is reported (Pintilie and Chiu, 2012[47]) to be faster than other fitting methods, such as ADP_EM, SITUS, Foldhunter and EMFit.
With the current advancements of electron microscopes and computational tools, subnanometer resolution (5-10 Å) structures are now easily obtainable. At these resolutions secondary structure elements (SSE) can be identified with help of component crystal structures or comparative models and the secondary structure elements α-helices and β-sheets appear as straight rods and curved plates respectively and loops as curved rods (Abeysinghe et al., 2008[1]; Ludtke et al., 2008[40]). Helixhunter and foldhunter are the computational methods which facilitate the quantitative identification of structural features in terms of known folds in three-dimensional density maps at different resolutions. Helixhunter is used to analyze a three-dimensional map for alpha helix content at intermediate resolutions, while foldhunter can be used to localize known or predicted folds or domains within larger macromolecular assemblies at lower resolutions (< 20 Å). Helixhunter and foldhunter are available within EMAN package (Jiang et al., 2001[31]). SSEHunter is also used for detection of alpha helices and beta sheets in subnanometer resolution Cryo-EM structures; SSEBuilder is a tool for the rapid identification and annotation of SSEHunter results. These are packaged within EMAN (Baker et al., 2007[5]). UCSF Chimera also includes these tools under analysis of intermediate resolution structures (AIRS) toolkit (Goddard et al., 2007[24]) (to which support is currently discontinued). Another recent tool for analyzing protein structures at near atomic resolution is Gorgon (Baker et al., 2011[4]), which is aimed at de novo model building in cryo-EM with the help of density skeletonization, SSE identification, building and correspondence, Cα placement and model optimization. The above mentioned SSEHunter is also incorporated into Gorgon to identify SSEs, but it includes more features than the EMAN version of SSEHunter.
For identifying conserved regions in cryo-EM maps of large macromolecular assemblies one can use MOTIF-EM (Saha et al., 2010[57]). SPI-EM can be used for predicting CATH superfamilies in 3D-EM Maps (Velazquez-Muriel et al., 2005[71]).
Apart from these techniques, Russel et al. (2012[56]) introduced a new method called the integrative modeling platform (IMP), a software package which combines the data from various individual experimental methods like X-ray crystallography, NMR spectroscopy, electron microscopy, small angle X-ray scattering etc. to generate new models and to overcome the barriers of each individual techniques. The modular structure of the 26S proteasome holocomplex is a recent example of the use of integrative approach (Lasker et al., 2012[38]).
Some of the recently studied structures by three dimensional electron microscopy and single particle analysis
26S proteasome: One of the recent structures solved by 3DTEM is that of 26S proteasome (2.5 MDa) from Saccharomyces cerevisiae, at a resolution of 7.4 Å (Fourier-Shell Correlation cut-off of 0.5). The molecular machine of the 26S proteasome is built from 31 different subunits, which catalyzes protein degradation. This high resolution structure was generated from 2.4 million individual particles. This map was used in conjunction with molecular dynamics-based flexible fitting to build a near-atomic resolution model of the holocomplex. They also determined the architecture of the lid complex subunits Rpn8/Rpn11. The map was released to the EM databank in 2012 (Beck et al., 2012[8]).
Native LDL particles: Low-density lipoprotein (LDL) particles are the major carriers of cholesterol in the human circulation. Kumar et al. (2011[36]) studied the three-dimensional structure of native LDL particles at 16 Å resolution at physiological human body temperature (37 oC). In the single particle reconstruction of LDL particles 52 micrographs at 6 oC and 23 micrographs at 37 oC were used, from which datasets of 71,521 and 29,844 images were resulted at 6 oC and 37 oC respectively and they found some noticeable differences like, the compact molecular packing of the core and order in a lipid-binding domain of apoB-100 were observed at 6 oC, but not at 37 oC and the features in the LDL particles that were not clearly separable in 3D maps at 6 oC, they were able to highlight them at 37 oC. They achieved a resolution of 16 Å for the final reconstructions of LDL at both temperatures using a Fourier Shell Correlation cutoff of 0.5. The density map was released in EM databank with an EMD id 2180.
Turnip crinkle virus: Bakker et al. (2012[7]) studied the 3D structures of both native and expanded forms of turnip crinkle virus (TCV), using cryo-electron microscopy, to visualize the encapsulated single-stranded RNA and coat protein (CP) N-terminal regions not seen in the high-resolution X-ray structure of the virion. Fitting studies were done with the crystal structure of the TCV S and P domains. To study the expansion in TCV, the native virions were incubated in low-ionic-strength buffer containing EDTA at pH 8.5. The resulted particles were used to determine an icosahedrally averaged, 3-D reconstruction for the expanded TCV virion at ∼17 Å resolution. The cryo-EM density maps for both native (11.5 Å) and expanded (17Å) TCVs determined by FSC at 0.5 cut-off were submitted to EM databank on with EMD ids 1863 and 1864 respectively.
Summary
As we have seen so far, three dimensional electron microscopy stands as an outstanding tool for macromolecular structure determination for molecules like membrane proteins and heterogeneous assemblies, apart from other conventional techniques. These studies are very much important to understand the structure-function relationships of the macromolecules. It also has a unique feature like visualization of structures in their native state. Though there is a resolution limit for structures achieved by these techniques, sub-nanometer resolution is now easily achievable on mid-range microscopes (Cong and Ludtke, 2010[14]). With further improvements of data acquisition and processing techniques and in silico tools one can have high resolution and more informative structure in near future.
Acknowledgement
Ayaluru Murali thanks University Grants Commission, New Delhi (India) for financial support.
References
[*] Corresponding Author:
Dr. Ayaluru Murali, Centre for Bioinformatics, School of Life Sciences, Pondicherry University, Puducherry-605014, India, eMail: murali@bicpu.edu.in