Research article
Anti-apolipoprotein A-I antibodies and paraoxonase 1 activity in Systemic Lupus Erythematosus
Mohammed Mahmoud Ahmed1[*], Eman Mahmoud Elserougy2, Iman Ibrahim Al-Gazzar2, Iman Mohamed Fikry1, Dawoud Fakhry Habib3, Khaled Mohamed Younes1, Neveen Abd El-hameed Salem4
1Internal Medicine Department, National Research Centre, Cairo, Egypt2Rheumatology and Rehabilitation Department, Cairo University, Cairo, Egypt
3Medical Biochemistry Department, National Research Centre, Cairo, Egypt
4Toxicology and Narcotics Department, National Research Centre, Cairo, Egypt
EXCLI J 2013;12:Doc719
Abstract
Systemic lupus erythematosus (SLE) patients have an increased risk of atherosclerosis. Identification of at-risk patients and the pathogenesis of atherosclerosis in SLE remain elusive. Paraoxonase 1 (PON1) and anti-apolipoprotein A-I antibody (anti-Apo A-I) appear to have a potential role in premature atherosclerosis in SLE. The aim of this work was to study PON1 activity and anti-Apo A-I antibody in SLE female patients and to demonstrate their relations to disease activity as well as disease related damage. Forty SLE female patients and 40 apparently healthy volunteers were included. Anti-Apo A-I antibodies levels and PON1 activity levels were assessed. Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) and systemic Lupus International Collaboration Clinics (SLICC)/American College of Rheumatology (ACR) damage index were preformed in all patients. Compared with controls, SLE patients showed significantly lower PON1 activity and significantly higher titers of anti-Apo A-I. Anti-Apo A-I antibody titers correlated inversely with PON1 activity. Elevated titers of anti-Apo A-I antibody and reduced PON activity were related to increased SLEDAI and (SLICC/ACR) damage index scores. We concluded that there is decreased PON1 activity and formation of anti-Apo A-I antibodies in female patients with SLE. SLE-disease activity assessed by SLEDAI and SLE disease related organ damage assessed by SLICC/ACR damage index are negatively correlated with PON1 activity and positively correlated with anti-Apo A-I antibodies. PON1 activity and anti-Apo A-I antibodies might be involved in the pathogenesis of atherosclerosis in SLE patients.
Keywords: SLE, anti-apolipoprotein A-I antibodies, paraoxonase 1
Introduction
Systemic lupus erythematosus (SLE) is a chronic inflammatory disorder characterized by the production of autoantibodies against a variety of self antigens. The clinical presentation of SLE varies widely with periodic flare-ups (Chiu et al., 2012[9]). Despite improvement in survival rates of patients, the standardized mortality ratios are still 3-fold higher than for the general population (Souza et al., 2012[41]).
Patients with SLE have a significantly increased risk of cardiovascular diseases (CVD) and mortality, particularly related to premature atherosclerosis. The exact etiology of premature atherosclerosis in SLE remains unclear. Traditional cardiac risk factors as defined by the Framingham Heart Studies, such as hypertension, hyperlipidemia, smoking, obesity and DM appear to play a critical role in the development of atherosclerosis, but these factors alone cannot adequately explain the increased incidence of atherosclerotic CVD in SLE. Nowadays, it is widely accepted that accelerated atherosclerosis in SLE is of a complex cross-talk between traditional and non-traditional SLE-related risk factors (Belibou et al., 2012[5]).
Systemic lupus erythematosus-related risk factors contributing to CVD are attributed to long-term inflammatory burden and immune abnormalities including innate immune responses, immune-cell activation, autoantibodies and increased levels of pro-inflammatory cytokines. In addition, oxidative stress, adipokines, dysfunctional lipids and multiple SLE therapeutics appear to affect the development and progression of atherosclerosis (Skaggs et al., 2012[40]).
Accumulating evidence has shown that atherosclerosis is not just a cholesterol storage disorder in vasculature but a sustained, dynamic, and chronic inflammatory process (Chiu et al., 2012[9]). Altered immune system function is recognized as the primary contributor to both the initiation and progression of atherosclerosis (Skaggs et al., 2012[40]).
Epidemiological studies have consistently shown that plasma HDL inversely correlated with the incidence of CVD in the general population. This relationship is actually quite complex and involves not only the quantity, but also the quality of HDL. The importance of HDL as a protective molecule against atherogenesis could be partly explained by its constituents mainly, paraoxonase (PON1) and apolipoprotein A-I (Apo A-I) (Guo et al., 2012[17]).
PON1is an enzyme synthesized in the liver and is bounded to HDL particles in blood. PON1 appears to contribute to the antioxidant and anti-atherosclerotic capabilities of HDL (Quillen et al., 2012[35]). However, the exact mechanism of PON1's protective action and its endogenous substrate remain elusive (Gugliucci et al., 2012[16]).
Apo A-I is the major protein component of HDL and is widely considered to be responsible for the anti-atherogenic and anti-thrombotic effects of HDL by promoting cellular cholesterol efflux and exerting anti-oxidative and anti-inflammatory effects (Eren et al., 2012[13]). Apo A-I exerts anti-oxidant properties by stabilizing PON1 (Narshi et al., 2011[30]). There is an emerging evidence of the presence of anti-Apo A-I antibodies and the reduction in the plasma levels of PON1 in SLE patients, thus interfering with the protective functions of HDL favoring atherogenesis (Batuca et al., 2009[3]).
The aim of this work was to study PON1 activity and anti-Apo A-I antibody in SLE female patients and to demonstrate their relations to disease activity as well as disease related damage.
Materials and Methods
Subjects
Eighty participants were included; they were divided into two groups.
Patient Group: Forty SLE premenopausal female patients diagnosed according to the ACR revised criteria of SLE (Hochberg, 1997[20]). Their age ranged between 18 to 46 years and the SLE disease duration extended between 0.5 and 15 years. Patients were selected from attendants of Rheumatology and Rehabilitation outpatient clinic and inpatient department of Kasr El-Aini hospital (Cairo University). Factors known to influence PON1 activity or induce premature atherosclerosis were excluded namely, smoking, diabetes mellitus, chronic renal failure, nephrotic syndrome and antiphospholipid syndrome. Patients on lipid lowering drugs, known cases of primary dyslipidemia and hypothyroidism and family history of CVD were excluded too.
Control Group: Forty apparently healthy age and culture matched female volunteers served as control group. Their age ranged between 18 to 47 years.
An informed consent was obtained from all participants in the study, and the study was approved by the ethical committees of Cairo University and National Research Center.
Methods
All participants were subjected to thorough history taking and physical examination. Blood samples were withdrawn from each participant after fasting for 12-14 hours. The following were determined: complete blood count using coulter counter (T660), erythrocyte sedimentation rate by Westergren method (Dacie and Lewis, 1985[10]), serum AST and ALT, blood urea and serum creatinine by automated Hitachi 911, urine analysis including: proteinuria, red blood cells, white blood cells, crystals and casts, serum lipid profile: HDL (Burstein et al., 1970[7]), triglycerides, total cholesterol (Jacobs and VanDemark, 1960[21]; Richmond, 1973[36]) and LDL concentration was estimated indirectly using the Friedewald equation: LDL = TC - HDL - (TG / 5) (Rifai and Warnick, 2006[37]), estimation of total albumin in 24 hours urine, anti-nuclear antibodies and anti-dsDNA antibodies by indirect immunoflourescence technique (Aitcheston and Tan, 1982[1]; Nakamura et al., 1984[29]), serum complement level (C3 and C4) quantitatively (Harris et al., 1986[19]) and Lupus anticoagulants (Thiagarajan et al., 1986[43]).
Determination of anti-Apo A-I antibodies level in the plasma
96-well plates (PolySorp) were half-coated for 1 hour at 37 °C with 10 mg/ml human Apo A-I (Sigma-Aldrich) in 70 % ethanol. Blocking was performed using phosphate buffered saline containing 1 % albumin from bovine serum for 1 hour at 37 °C. A hundred microlitres of the samples (1:300 dilutions in blocking agent) and positive control were added to duplicate wells in both halves of the plate for 1 hour at 37 °C. After washing, alkaline phosphatase-conjugated anti-human IgG (1:1000 in the blocking agent) was added for 1 hour. p-Nitrophenyl phosphate (1:5000 in bicarbonate buffer, pH 9.8) was added and incubated at 37 °C for color development and the absorbance read at 405 nm after 1 hour. All assays were validated by the inclusion of internal quality control samples of known activity. The results were expressed as the percentage of the positive control present in each plate after subtraction from the background in the uncoated half of the plate. Inter-/intra-plate coefficients of variation were < 10 % (Batuca et al., 2009[3]).
Determination of paraoxonase1 activity
Serum paraoxonase 1/Arylesterase (the activity toward phenyl acetate) was determined spectrophotometrically at 270 nm with phenyl acetate used as the substrate. The assay mixture included 1.0 mmol/L of phenyl acetate and 0.9 mmol/L CaCl2 in 20 mmol/L Tris HCl, pH 8.0 at 25 °C. Nonenzymatic hydrolysis of phenyl acetate was subtracted from the total rate of hydrolysis. The E270 for the reaction is 1310 mol/L-1 cm-1. The results are expressed in U/ml; 1 U hydrolyzes 1 µmol of phenyl acetate/minute (Aviram et al., 2000[2]).
Assessment of disease activity and disease related damage
Assessment of disease activity was done using Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) (Bombardier et al., 1992[6]). Assessment of disease related damage was done using using Systemic Lupus International Collaborative Clinics/American College of Rheumatology (SLEICC/ACR) Damage Index (Gladman et al., 1996[15]).
Statistical analysis
Data were statistically described in terms of mean standard deviation ( SD). Comparison between cases and control groups was done using Student t test for independent samples. Correlation between various variables was done using Pearson moment correlation equation for linear relation in normally distributed variables and Spearman rank correlation equation for non-normal variables. p values less than 0.05 was considered statistically significant. All statistical calculations were done using computer programs SPSS (Statistical Package for the Social Science; SPSS Inc., Chicago, IL, USA) version 15 for Microsoft Windows.
Results
The statistical analyses of the demographic and laboratory characteristics of the participants, of the main clinical data of SLE patients and of the drugs given to them are shown in Tables 1(Tab. 1), 2(Tab. 2) and 3(Tab. 3) respectively.
The mean serum Paraoxonase 1 (PON1) activity level in SLE patients was significantly lower than its mean level in the control. The mean plasma anti-Apo A-I antibody level in SLE patients was significantly higher than its mean level in the controls (Table 4(Tab. 4)). A significant negative correlation was obtained between the serum PON1 activity level and the plasma anti-Apo A-I level in SLE patient (Figure 1(Fig. 1)).
The correlative studies between SLEDAI scores and PON1 activity as well as anti-Apo A-I antibody showed a marked significant negative correlation between SLEDAI scores and PON1 and a significant positive correlation with the anti-Apo A-I antibody. The correlative studies also showed a marked significant negative correlation between SLICC/ACR scores and PON1 activity, whereas a significant positive correlation with the anti-Apo A-I antibody was obtained (Table 5(Tab. 5)).
Non significant correlations were obtained between both PON1 and anti-Apo A-I levels in SLE patient and the demographic characteristics and the laboratory investigations of these patients. However, significant negative correlations were obtained between C3, C4 and anti-Apo A-I antibodies levels but not with PON1 activity (Table 6(Tab. 6)). Again, non significant correlations were obtained between both PON1 activity and anti-Apo A-I of the patients and the dose and the duration of the drug used (Table 7(Tab. 7)).
Statistically significant differences were present between PON1 activity levels in the presence and absence of constitutional and CNS manifestations. Otherwise, no significant differences were present between PON1 activity levels in the presence and absence of other disease manifestations (Table 8(Tab. 8)). Statistically non-significant differences were present between the Anti-Apo A-I levels in the presence or absence of all the studied disease manifestations (Table 9(Tab. 9)).
Discussion
Our study revealed statistically significant lower plasma PON1 activity in SLE patients compared to the healthy controls. Thus, our patients are prone to atherosclerosis and its complications. There are increasing epidemiological evidences that PON1 protects against development of atherosclerosis and is an independent risk factor for CVD (Fuhrman, 2012[14]). In addition, decreased PON1 activity is associated with clinical atherothrombotic complications (Kiss et al., 2007[24]). Furthermore, the atheroprotective function of PON1 has been also demonstrated in PON1 knock-out mice, which exhibited an accelerated atherosclerosis (Marchegiani et al., 2012[26]).
The decreased PON1 activity in our patients may be a cause or a consequence of SLE. This issue remains unclear. As a cause, Tripi and his coworkers in (2006[44]) concluded that low PON1 activity is independently associated with SLE. On the contrary, reduced PON1 activity could be a consequence of inflammation and oxidative stress (Batuca et al., 2009[3]).
Assessment of PON1 activity in SLE patients showed conflicting results. Most studies reported decline of PON1 activity (Delgado Alves et al., 2002[12]; Batuca et al., 2007[4], 2009[3]). The investigators assessed PON1 activity by paraoxon as a substrate in their assay. On the other hand, on using both phenyl acetate and paraoxon as substrates in the assay in the same SLE patients (Tripi et al., 2006[44]; Kiss et al., 2007[24]), there was significant decline in PON1 activity when assessed by paraoxon. However, on assessment of PON1 activity by phenyl acetate, there was significant increase in PON1 activity in a study (Tripi et al., 2006[44]), whereas, there was no significant change in PON1 activity in the other study. In our work we used phenyl acetate as a substrate for assessment of PON1 activity.
These contrasting results of PON1 activity levels in SLE may be attributed to the fact that PON1 activity differs greatly among ethnic groups. Inter-individual variation in PON1 activity is under strong genetic influence (Dasgupta et al., 2011[11]) and pharmacological agents can also modulate PON1 activity (Precourt et al., 2011[33]). On the other hand, the difference in the results of PON1 activity levels in the same patients may be attributed to genotype variation and polymorphism (Tripi et al., 2006[44]; Kiss et al., 2007[24]). Polymorphisms can generate opposite effects on PON1 activity depending on type of substrate used (Precourt et al., 2011[33]).
Using phenyl acetate in our study as a substrate minimizes the role of genotype variation (Precourt et al., 2011[33]). The difference between our results and the results of those who used phenyl acetate also in their studies (Tripi et al., 2006[44]; Kiss et al., 2007[24]) appears to be due to the exclusion of the individuals taking statins from our study. Statins has in general been associated with an increase in the PON1 serum activity measures (Camps et al., 2012[8]).
In our study, PON1 activity showed statistically significant negative correlation with SLE disease activity assessed by SLEDAI scores. Similar result was obtained between PON1 activity and British Isles Lupus Assessment Group (BILAG-2004) disease activity index. It has been reported that SLE activity is associated with deterioration of antioxidant status (Batuca et al., 2009[3]). Increased oxidative stress inactivates PON1, and the decline in PON1 activity in turn augments oxidative stress. Therefore, the trio of disease activity, oxidative stress, and PON1 activity forms a vicious circle (Camps et al., 2012[8]; Li et al., 2012[25]) and this explains the negative correlation between PON1 activity and SLEDAI.
In contrast to our results, Kiss and his coworkers in 2007[24] found no correlation between PON1 activity and SLEDAI. This may be attributed to the relatively lower SLEDAI scores of the patients included in their study (2) versus (7.9) in our study.
There was a statistically significant negative correlation between SLICC/ACR damage index scores and PON1 activity in our study. Our data is consistent with a previous report (Batuca et al., 2009[3]). The reported oxidative stress in SLE can damage macromolecules and can exacerbate inflammation leading to tissue damage (Shah et al., 2011[38]). This may be an explanation for this result.
In this study, statistically significant differences were obtained between the PON1 levels in the presence and absence of constitutional and CNS manifestations only. To our knowledge, this is the first study to address this relationship in SLE. On the other hand, Tripi et al., (2006[44]) found decreased (though non-significant) PON1 activity levels in lupus nephritis patients compared to those without lupus nephritis. To document possible relationships between PON1 activity and other clinical manifestations of SLE appears to need large samples as SLE is a heterogeneous disease.
Apolipoprotein A-I is an important component of HDL and it has cardioprotective effects (Eren et al., 2012[13]). Autoantibodies directed to Apo A-I molecules may inhibit their normal functions (Hahn, 2010[18]). These antibodies may be present in general population but in low titers and it may be related to the vascular and immune ageing processes (Batuca et al., 2007[4]).
In this study, there was a statistically significant higher anti-Apo A-I antibody levels in SLE patients as compared to the healthy controls. These antibodies have been identified in SLE (O’Neill et al., 2010[31]). However, there is paucity of information about its exact pathophysiological role (Hahn, 2010[18]). Anti-Apo A-I antibody production may be explained by the fact that autoantibodies production against multiple self antigens is considered as a hallmark of SLE (Joo et al., 2012[23]). In autoimmune diseases, oxidative stress is the major event causing structural modifications of proteins with consequent appearance of neoepitopes which can become targets of autoimmune reactions, thus sustaining the inflammatory mechanisms involved in endothelial dysfunction and plaque development (Profumo et al., 2011[34]).
These autoantibodies may lead to Apo A-I dysfunction. Apo A-I normally contributes in the removal of damaged and apoptotic endothelial cells. Thus, anti-Apo A-I antibodies that inhibit these normal functions could predispose to autoimmunity and may play a role in the pathogenesis of SLE. Even if these antibodies arise later, in the development of the disease, and even if disease is already established, these antibodies may promote a vicious cycle perpetuating the pathological process underlying the disease (Shoenfeld et al., 2007[39]; Hahn, 2010[18]).
Apolipoprotein A-I antibodies can affect atherosclerosis and thrombosis independent of autoimmunity (Hahn, 2010[18]). It could represent an emerging CVD risk factor and an independent predictor of major CVD events (Vuilleumier et al., 2010[45]). The presence of these antibodies in our patients might have exposed them to accelerated atherosclerosis as they lose their atheroprotection mediated by Apo A-I. These antibodies can also affect the normal atheroprotective function of HDL, and the formed dysfunctional HDLs are proinflammatory (Hahn, 2010[18]).
Our study revealed statistically significant lower C3 and C4 levels in SLE patients compared to the healthy controls. However, the mean values of C3 and C4 in the SLE patients and controls were within the normal range. Thus, there may be a mild degree of complement activation. Negative correlation was obtained between anti-Apo A-I antibody with C3 and C4 levels. Circulating immune complexes have been correlated with complement activation and autoantibody profiles in SLE (Mathsson et al., 2007[27]). This may be a potential explanation for the activation and consumption of C3 and C4 associated with the increase in production of autoantibodies against Apo A-I in our patients.
There was a significant positive correlation between anti-Apo A-I antibody and SLE disease activity assessed by SLEDAI scores in our patients. In SLE, levels of certain antibodies such as anti-dsDNA frequently correlate with the disease activity (O’Neill et al., 2010[31]). Similarly, other studies reported association between anti-Apo A-I antibody and SLE disease activity using other different scoring systems than SLEDAI. Significant positive correlation between anti-Apo A-I antibody levels, and SLE disease activity assessed both by BILAG (Batuca et al., 2007[4]) and by the European Consensus Lupus Activity Measurement (ECLAM) in their German patients (Shoenfeld et al., 2007[39]) have been reported.
There was a statistically significant positive correlation between SLICC/ACR scores and anti–Apo A-I antibody levels in our study. Our results confirm the findings of others (Batuca et al., 2009[3]). This can be explained by the formation of antibody–antigen complexes that can attach to tissues, fix complement, and cause its damage (Hahn, 2010[18]).
Compared to controls, our patients have dyslipidemia in the form of reduced levels of HDL and elevated levels of TC, TG, and LDL. This pattern of dyslipidemia is associated with adverse CVD risk in the general population (Ji et al., 2011[22]).
From the above discussion, our patients appear to be in a proatherogenic state. Traditional risk factors involved in the pathogenesis of atherosclerosis in our patients include dyslipidemia, corticosteroid administration and hypertension. SLE-related risk factors in our patients include: immune system involvement (anti-Apo A-I antibody production), and oxidative stress (reduced PON1 activity) (Skaggs et al., 2012[40]).
Increasing evidence has highlighted the role of oxidative stress in both SLE as well as atherosclerotic CVD. However, the molecular bases of the interaction between increased oxidative stress, SLE and atherosclerosis remain unclear (Li et al., 2012[25]). This study may provide a plausible explanation for these clues. PON1 physiologically plays an important role in decreasing oxidative stress and atherosclerosis risk. It accounts for most of the capacity of HDL to prevent oxidation of LDL (Fuhrman, 2012[14]). So, the reduced PON1 activity in our patients might have a deleterious effect on the protective function of HDL. SLE is associated with intense antibody production and a possible mechanism for the reduced antioxidant PON1 activity may be the autoantibody production against Apo A-I present in our patients (Srivastava et al., 2011[42]). The statistically significant negative correlation between PON1 and anti-Apo A-I antibody in the present study together with other research groups (Batuca et al., 2007[4], 2009[3]) highlighted this concept. Our results suggest the presence of a vicious circuit between PON1 activity and anti-Apo A-I antibody. Reduced PON1 activity affects HDL functions and this enhances the oxidative stress. Oxidative stress leads to the formation of new antigens which in turn enhances the humoral response leading to the production of autoantibodies including anti-Apo A-I antibodies. Anti-Apo A-I antibodies target PON1 reducing its activity (Srivastava et al., 2011[42]; Fuhrman, 2012[14]). Such a vicious circuit with its components maintains oxidative stress and subsequent SLE-related pro-atherogenic state.
Diminished HDL levels have been recognized as an independent risk factor for CVD. However, there is an increasing evidence that HDL quality may be as important as its quantity. The concept of "dysfunctional HDL" or "proinflammatory HDL" is an emerging issue. Dysfunctional HDL converts a positive force protecting arteries to a negative one, enhancing atherogenesis (Otocka-Kmiecik et al., 2012[32]). Among the factors that convert HDL into "dysfunctional HDL" is the decrease of the major antioxidant enzyme PON1. Another major structural and functional component of HDL is Apo A-I. Autoantibodies against Apo A-I may lead to Apo A-I dysfunction with secondary HDL dysfunction (McMahon, 2009[28]; Hahn, 2010[18]).
HDL may be affected in our SLE patients in the same manner. The two main components that accounts for most of the protective functions of HDL are affected. PON1 activity is diminished and Apo A-I are targeted by an anti-Apo A-I antibodies. So, our patients appear to have dysfunctional HDL. Dysfunctional HDL is another pro-atherogenic factor (McMahon et al., 2009[28]) in our SLE patients.
Finally, over the next few years, the two SLE related risk factors of atherosclerosis in this study anti-Apo A-I antibodies and/or PON1 activity may be recommended as a routine strategy to stratify patients for risk of accelerated atherosclerosis.
Conclusion
There is decreased PON1 activity and formation of anti-Apo A-I antibodies in female patients with SLE. SLE-disease activity assessed by SLEDAI and SLE disease related organ damage assessed by SLICC/ACR damage index are negatively correlated with PON1 activity and positively correlated with anti-Apo A-I antibodies. PON1 activity and anti-Apo A-I antibodies might be involved in the pathogenesis of atherosclerosis in SLE patients.
References
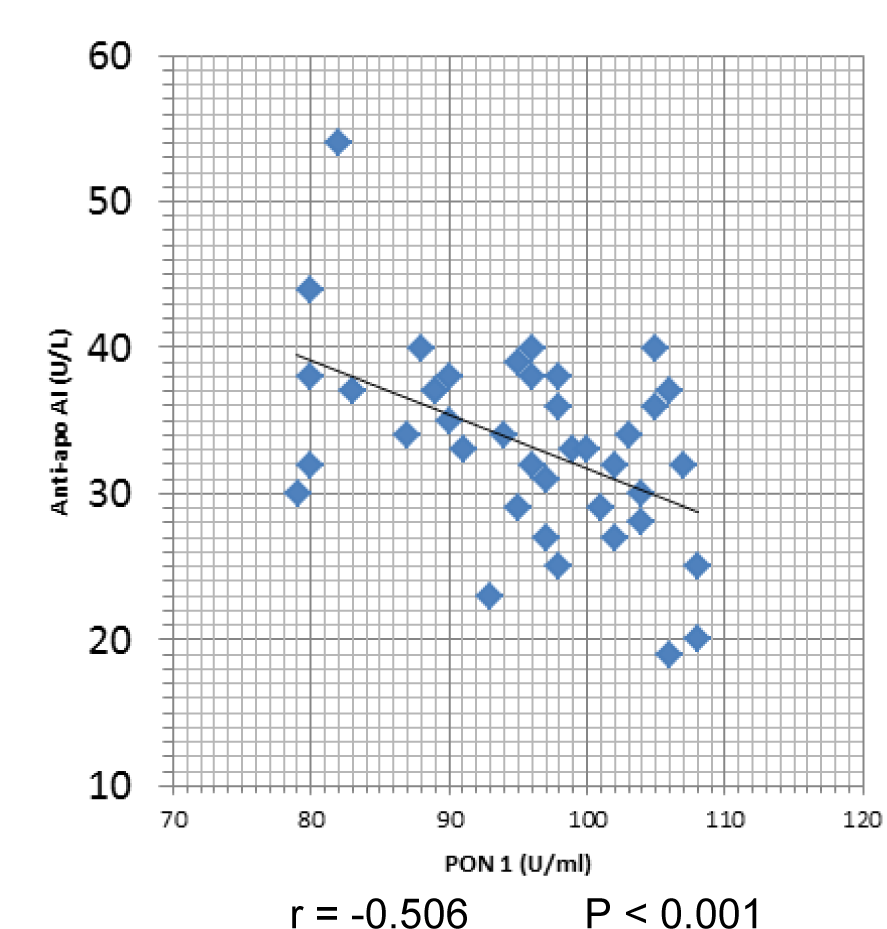
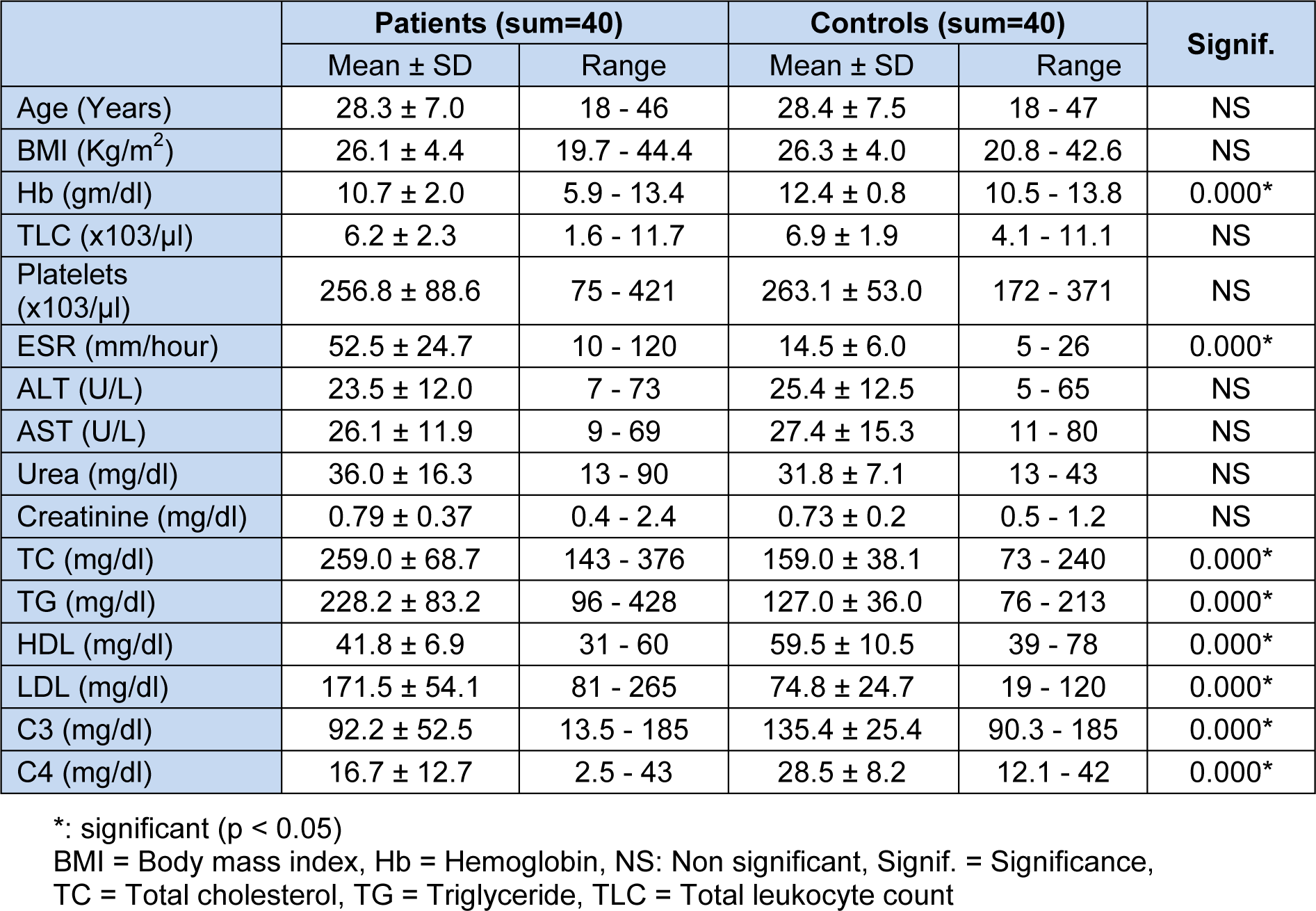
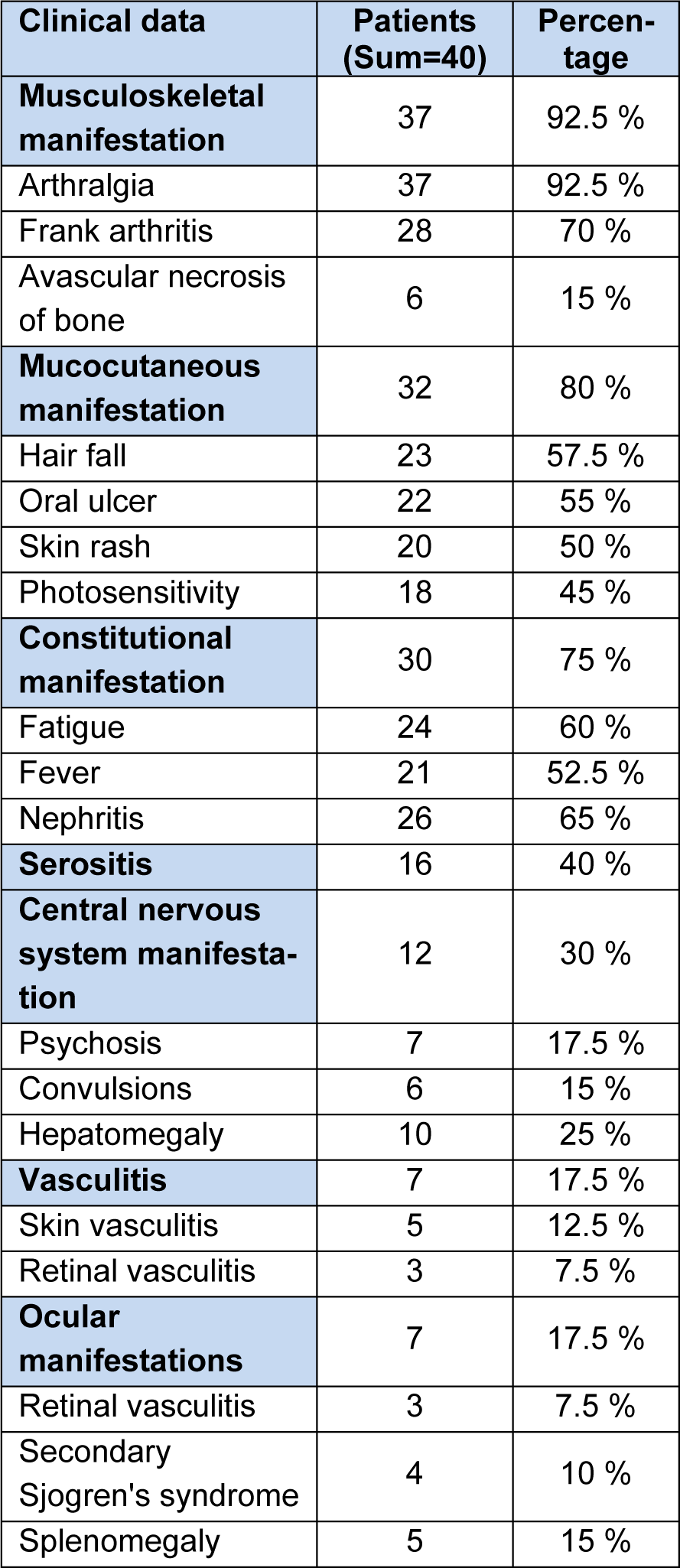
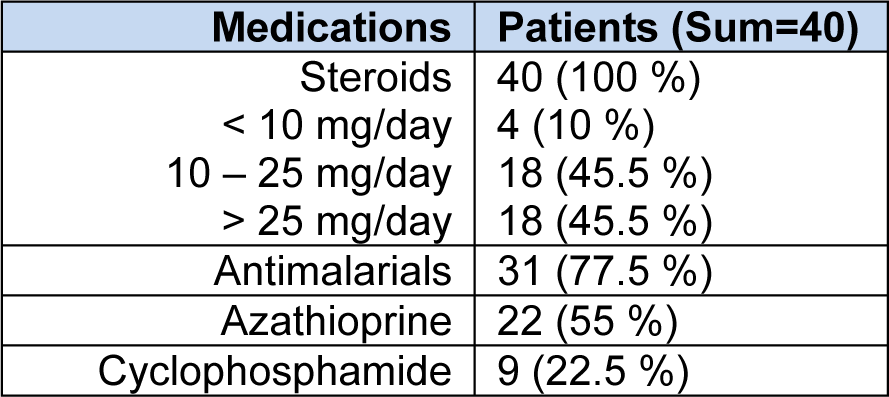
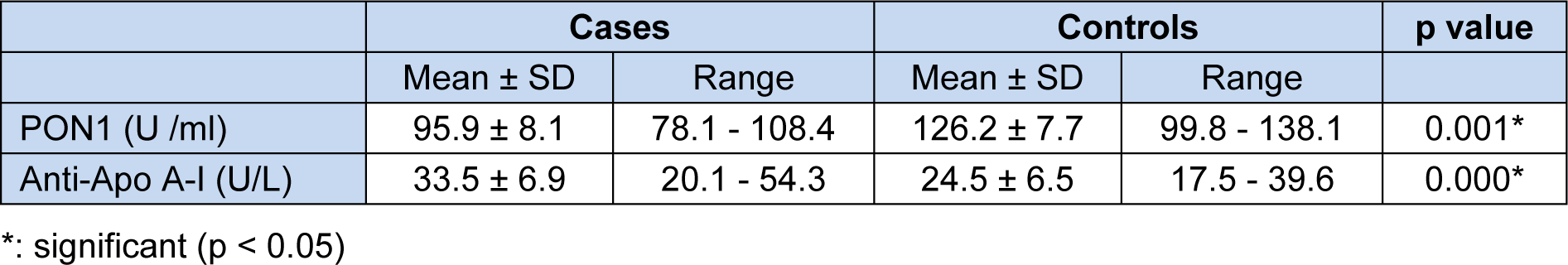
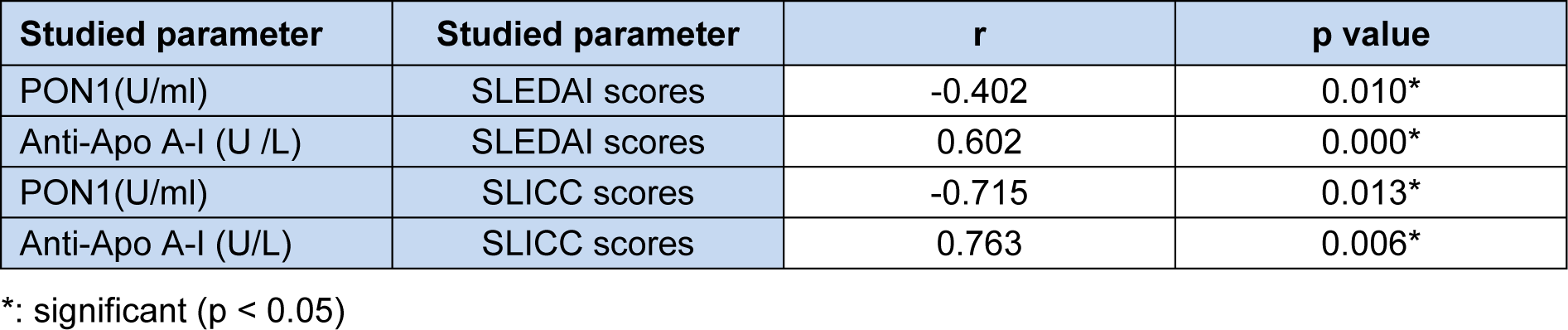
Table 5: Correlation between PON1 activity and Anti-Apo A-I antibody levels in SLE patients and the SLEDAI and SLICC scores
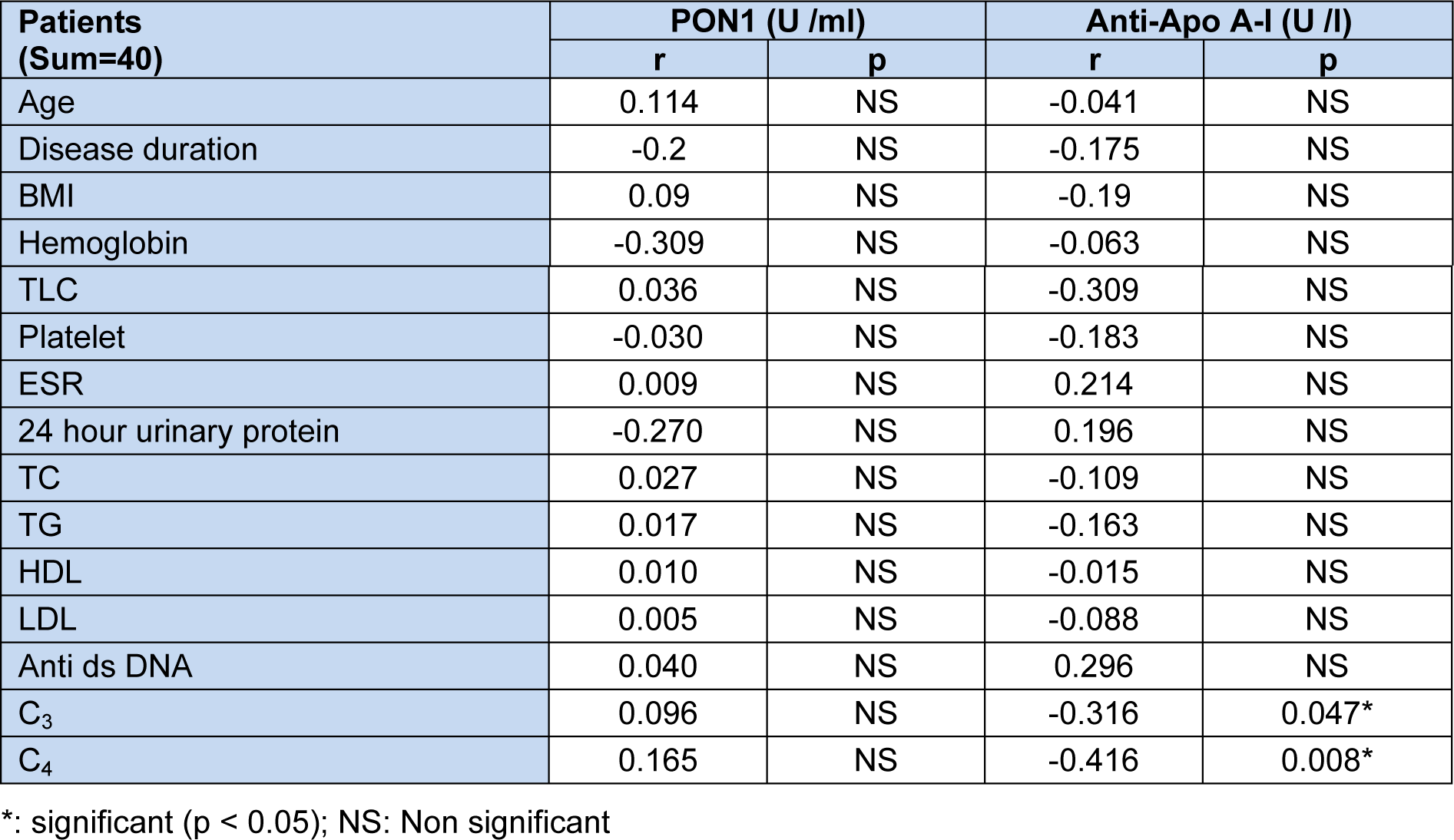
Table 6: Correlation between PON1 activity, anti-Apo A-I and the demographic characteristics and the laboratory investigations of the SLE patients
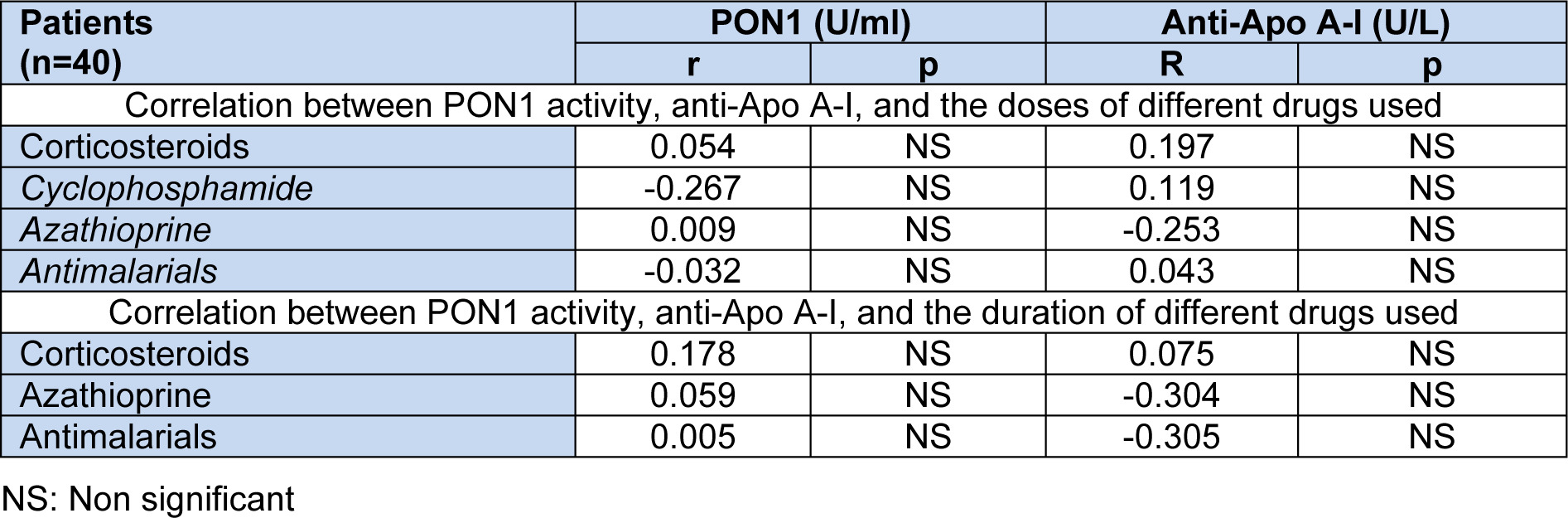
Table 7: Correlation between PON1 activity, anti-Apo A-I, the doses and the duration of different drugs used
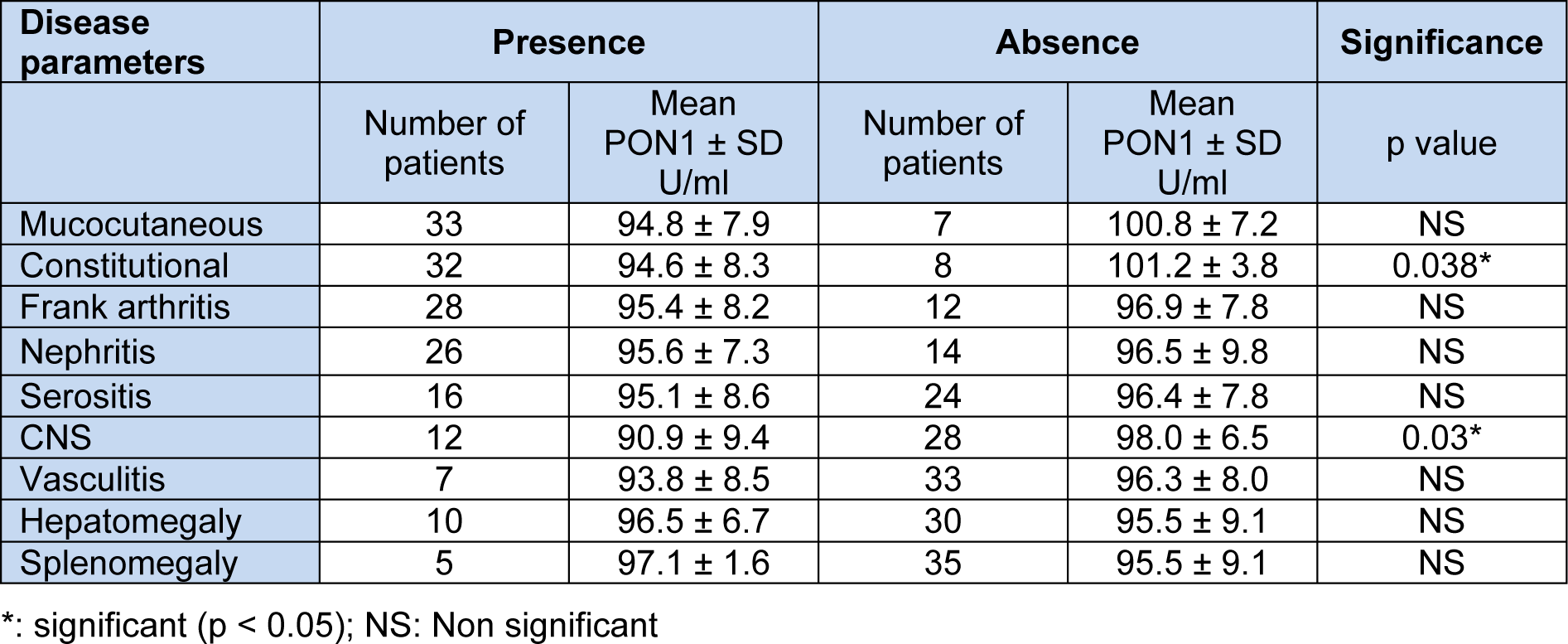
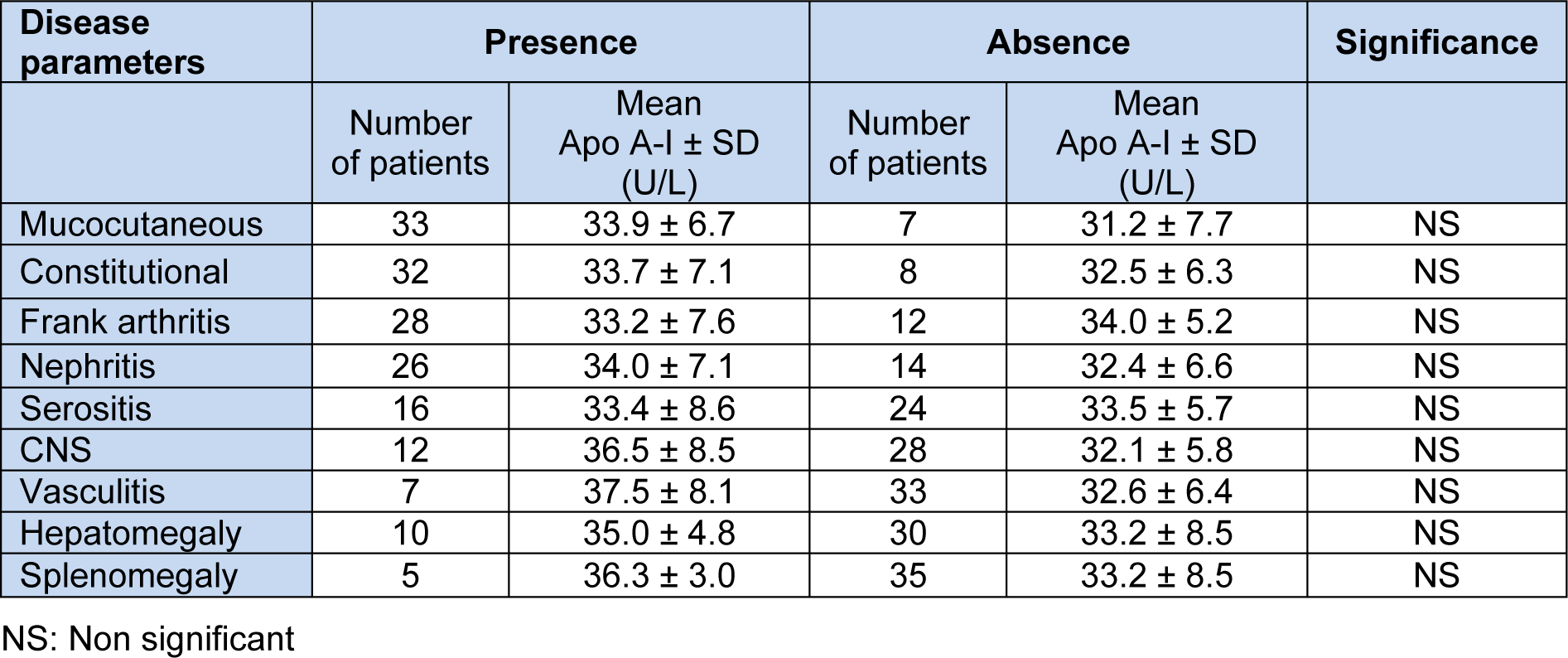
Table 9: Apolipoprotein A-I antibody levels in the presence and the absence of different disease manifestations
[*] Corresponding Author:
Mohammed Mahmoud Ahmed, National Research Centre, El Buhouth St., Dokki, Cairo, Egypt, Postal Code: 12311, eMail: drmohmah@yahoo.com