Research article
A simple click by click protocol to perform docking: AutoDock 4.2 made easy for non-bioinformaticians
Syed Mohd. Danish Rizvi1, Shazi Shakil2[*], Mohd. Haneef2
1Department of Biosciences, Integral University, Lucknow, India-2260262Department of Bio-engineering, Integral University, Lucknow, India-226026
EXCLI J 2013;12:Doc831
Abstract
Recently, bioinformatics has advanced to the level that it allows almost accurate prediction of molecular interactions that hold together a protein and a ligand in the bound state. For instance, the program AutoDock has been developed to provide a procedure for predicting the interaction of small molecules with macromolecular targets which can easily separate compounds with micromolar and nanomolar binding constants from those with millimolar binding constants and can often rank molecules with finer differences in affinity. AutoDock can be used to screen a variety of possible compounds, searching for new compounds with specific binding properties or testing a range of modifications of an existing compound. The present work is a detailed outline of the protocol to use AutoDock in a more user-friendly manner. The first step is to retrieve required Ligand and Target.pdb files from major databases. The second step is preparing PDBQT format files for Target and Ligand (Target.pdbqt, Ligand.pdbqt) and Grid and Docking Parameter file (a.gpf and a.dpf) using AutoDock 4.2. The third step is to perform molecular docking using Cygwin and finally the results are analyzed. With due confidence, this is our humble claim that a researcher with no previous background in bioinformatics research would be able to perform molecular docking using AutoDock 4.2 program by following stepwise guidelines given in this article.
Keywords: computer aided docking, free offline docking, non-bioinformaticians, AutoDock, drug discovery, enzyme-ligand interaction
Introduction
Computer-aided docking is an important tool for gaining understanding of the binding interactions between a ligand (small molecule) and its target receptor (enzyme) (Anderson, 2003[1]; Schneider, 2010[6]) and has emerged as a reliable, cost-effective and time-saving technique for the discovery of lead compounds (Walters et al., 1998[8]; Schneider and Böhm, 2002[7]; Waszkowycz et al., 2001[10]). In recent years, the virtual screening approach for docking small molecules into a known protein structure is a powerful tool for drug design and has become an integral part of the drug discovery process. Computational tools like AutoDock offer the advantage of delivering new drug candidates more quickly and at a lower cost (Gilbert, 2004[2]; Warren et al., 2006[9]). AutoDock is an excellent non-commercial docking program that is widely used. Further, it employs a stochastic Lamarckian genetic algorithm for computing ligand conformations and simultaneously minimizing its scoring function which approximates the thermodynamic stability of the ligand bound to the target protein (Morris et al., 1998[4], 2009[5]). The use of complementary experimental and informatics techniques increases the chance of success in many stages of the discovery process. Theoretically the application of AutoDock in virtual screening is constrained only by the chemical compounds features that can be calculated and the relation between these features and the target (Lazarova, 2008[3]). But the problem arises in practical implementation of AutoDock in virtual screening of compounds which requires several considerations. Thus, this paper provides an easier protocol for the use of AutoDock for molecular docking purposes and will hopefully help in practically implementing AutoDock and AutoDock tools for the virtual screening purposes. To make it easier to understand, an example of experiment of the docking of Imipenem-hydrolyzing enzyme beta-lactamase SME-1 with Imipenem as ligand was made using AutoDock 4.2/ADT.
Requirements
1. Windows XP or Windows 7
Freely available software’s for non-commercial uses:
2. MGL tools
http://mgltools.scripps.edu/downloads
3. Cygwin
http://www.cygwin.com/install.html
(Click setup-x86.exe for 32-bits version while setup-x86_64.exe for 64-bits version)
4. Discovery Studio Visualizer
http://accelrys.com/products/discovery-studio/visualization-download.php
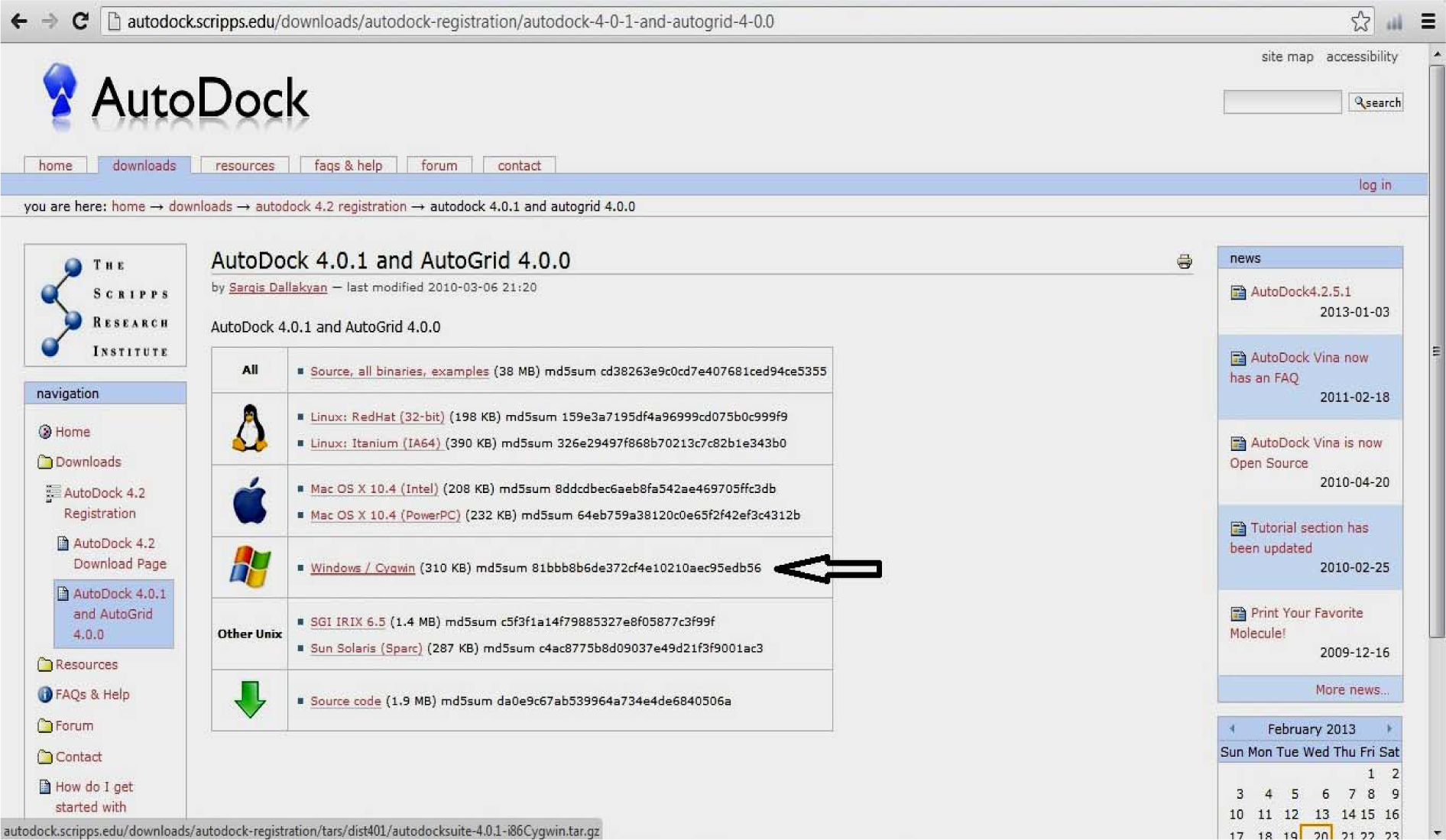
5. Binary files
http://autodock.scripps.edu/downloads/autodock-registration/autodock-4-0-1-and-autogrid-4-0.0
(Fig. 1)
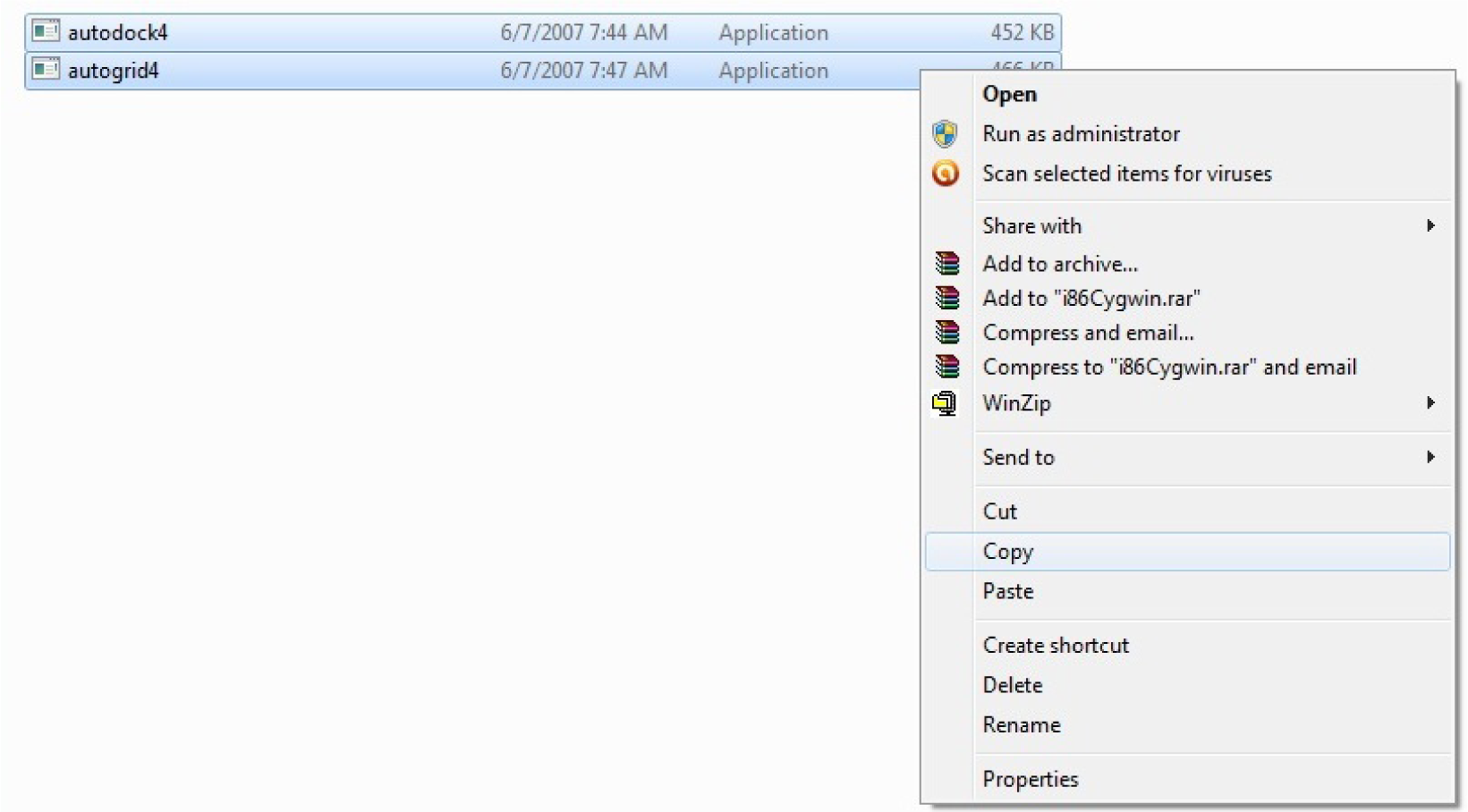
Download and Extract autodocksuite-4.0.1-i86Cygwin.tar
Copy autodock4.exe and autogrid4.exe
(Fig. 2)
Paste in My computer\ C drive\ Cygwin\ bin
6. Java
http://www.java.com/en/download/index.jsp
Methods
1 Retrieving Required Ligand and Target .pdb files from major databases:
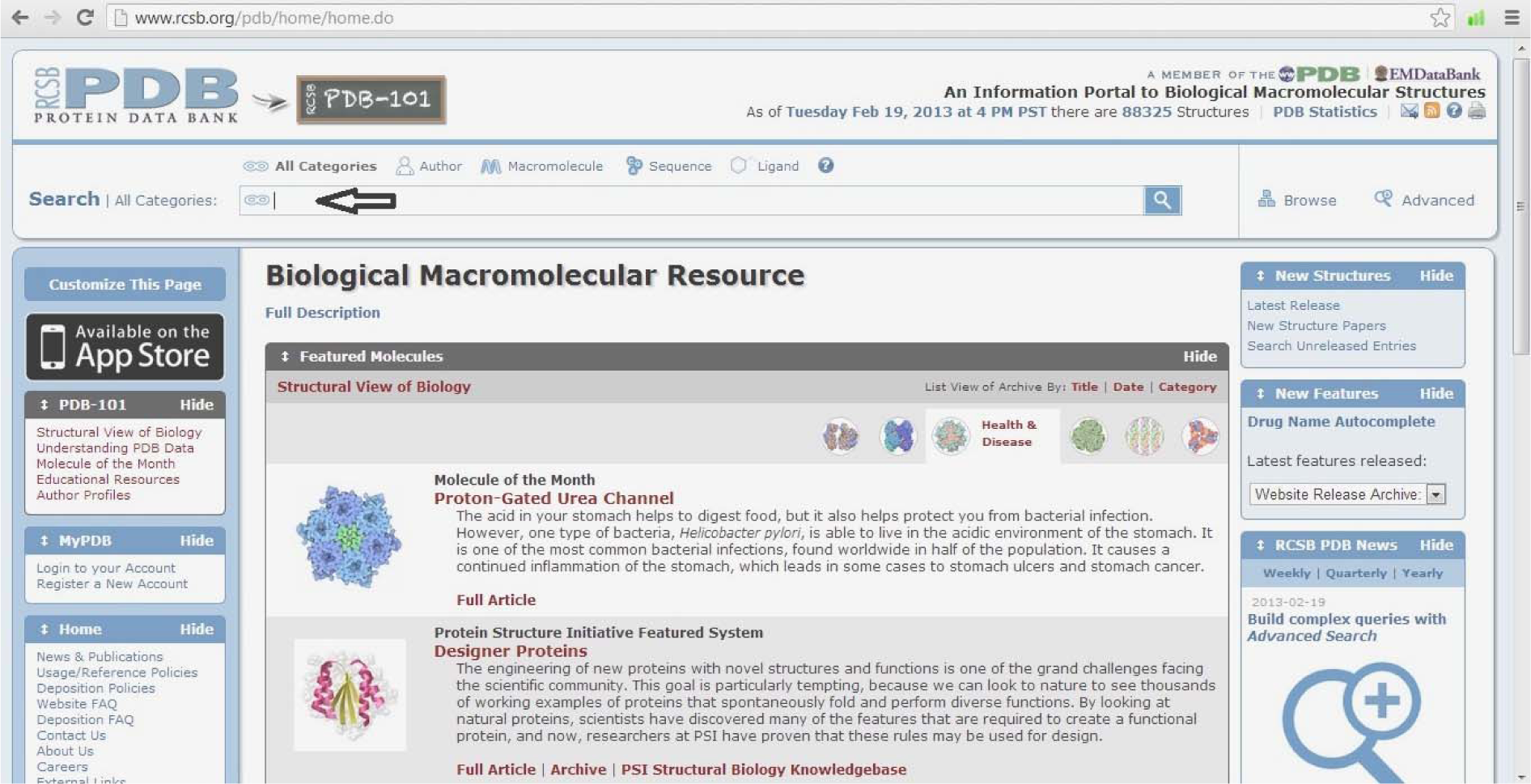
1.1 Retrieving Target.pdb files from major protein databases
http://www.rcsb.org/pdb/home/home.do
(Fig. 3)
- Type the query protein or enzyme (Imipenem-hydrolyzing beta-lactamase SME-1)
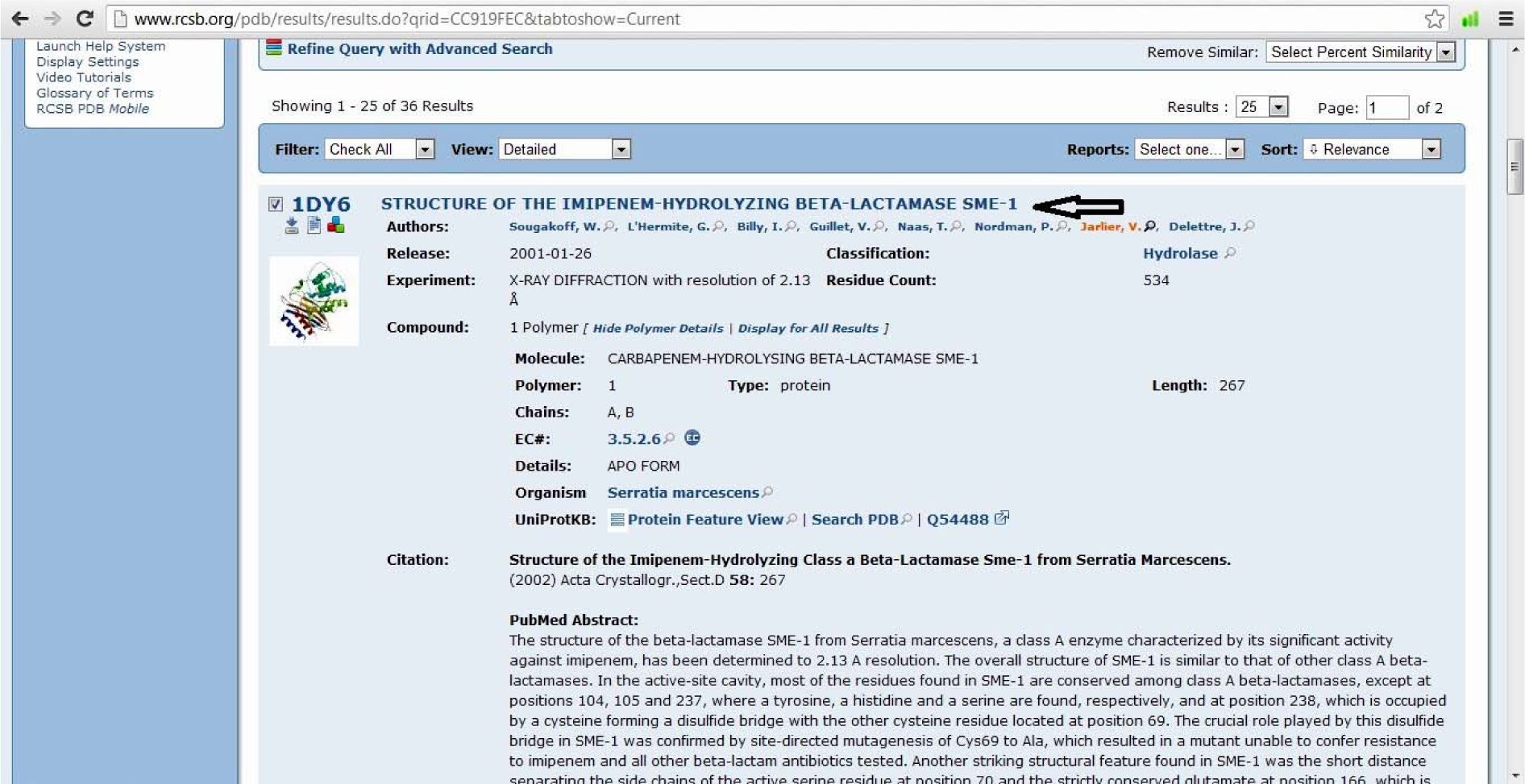
- Select enzyme (Imipenem-hydrolyzing beta-lactamase SME-1)
(Fig. 4)
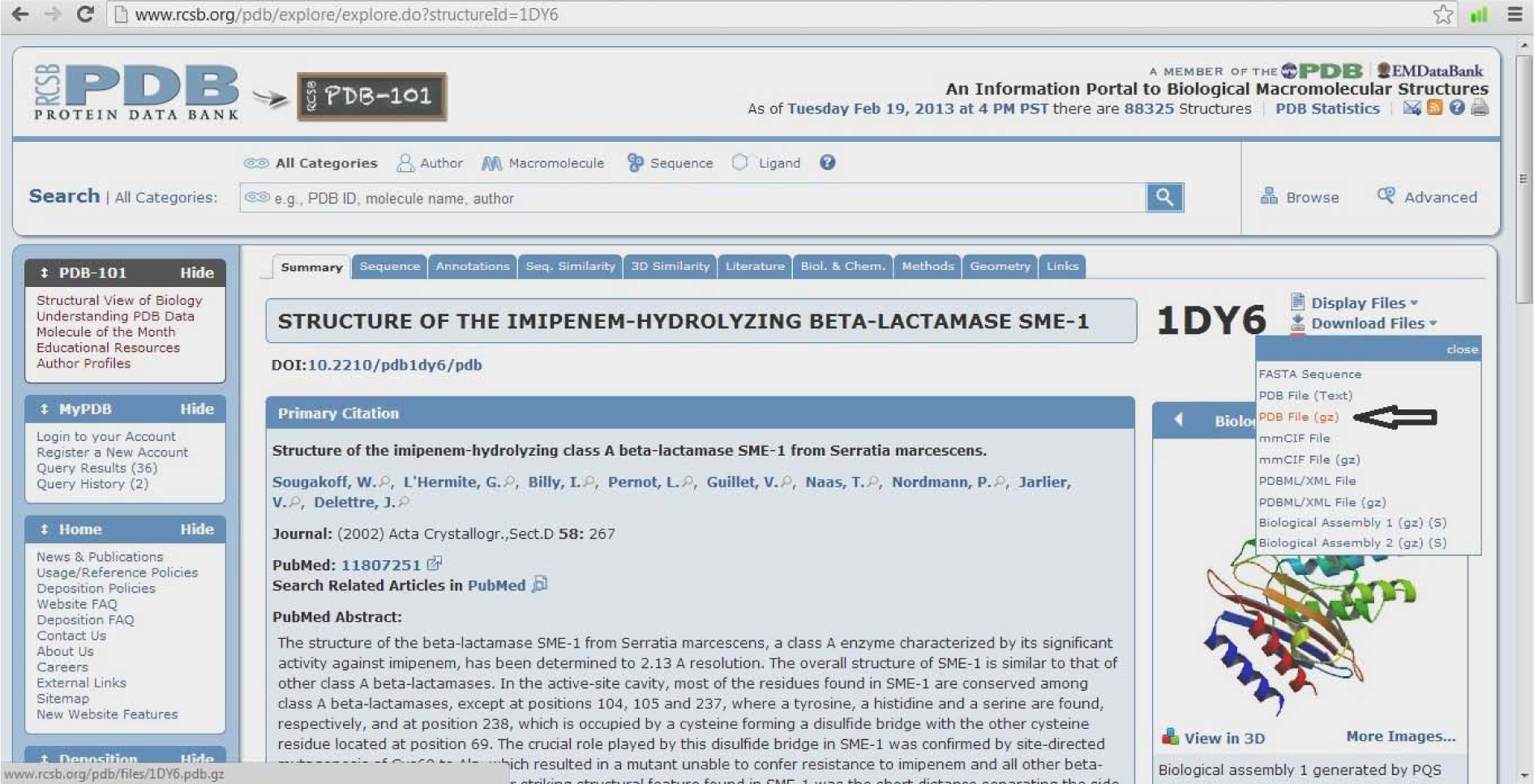
- Select download files
- Click PDB file (gz) and download it
(Fig. 5)
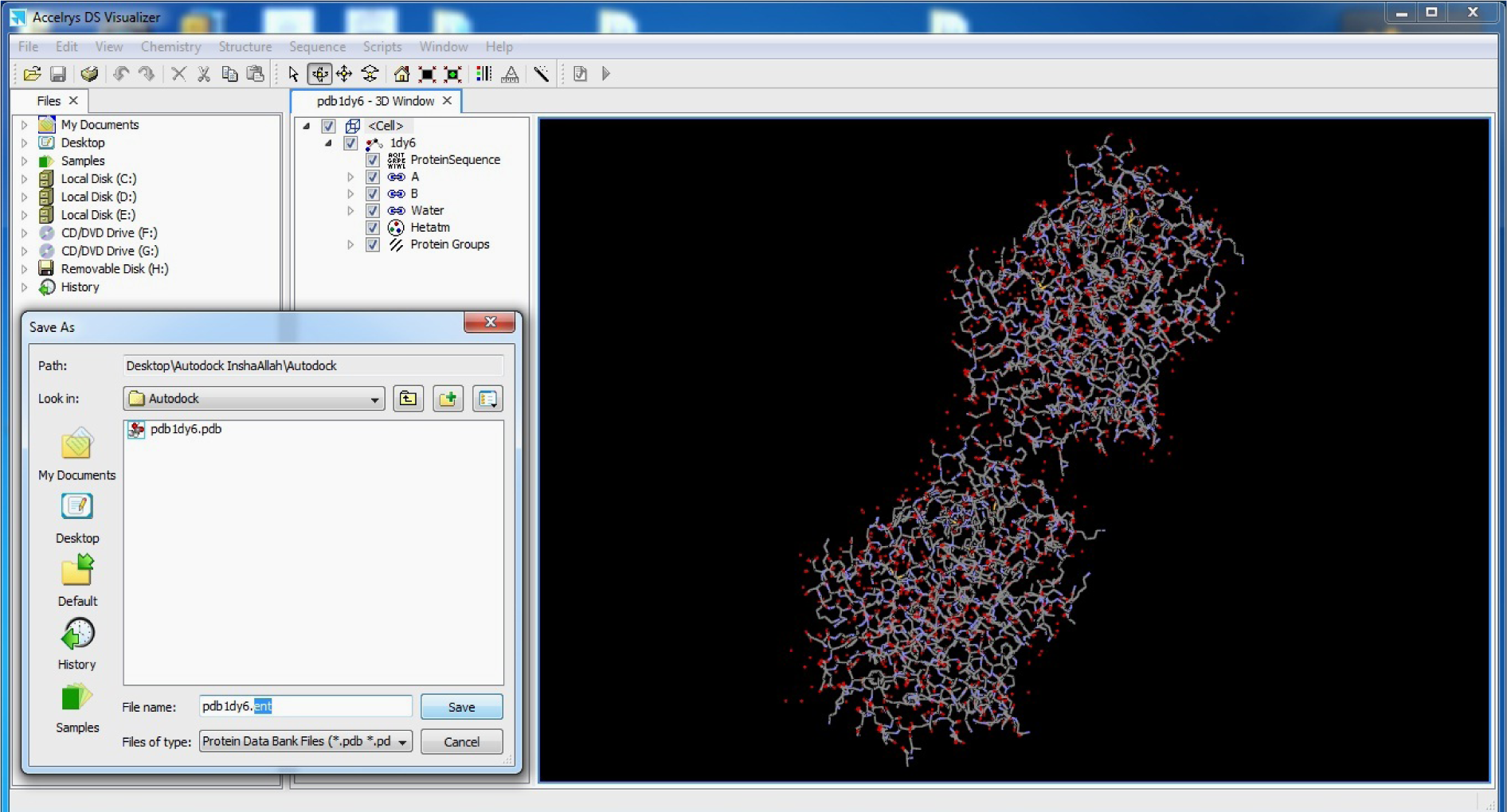
- Open it in Discovery Studio Visualiser
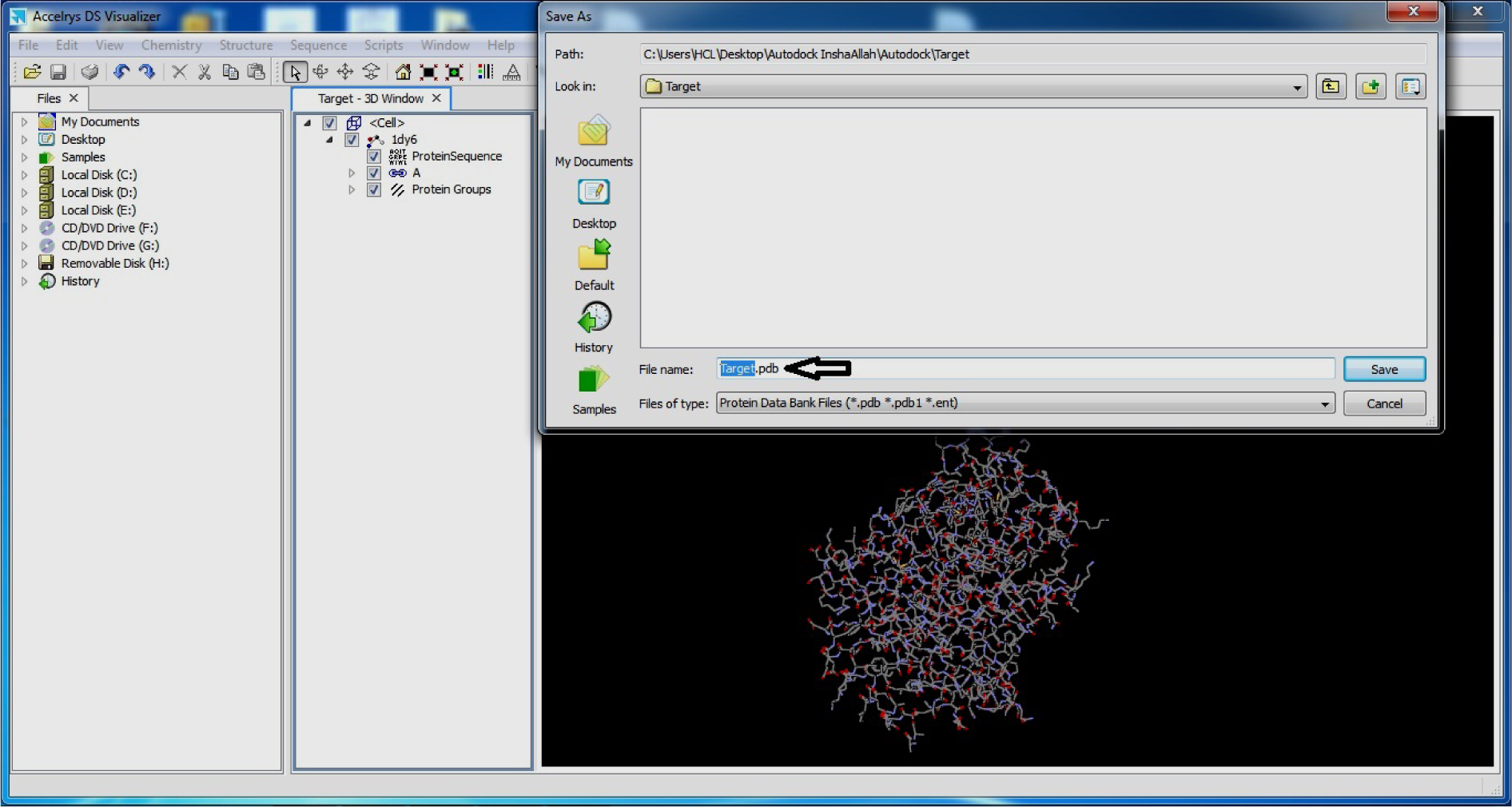
- Save as .pdb format
(Fig. 6)
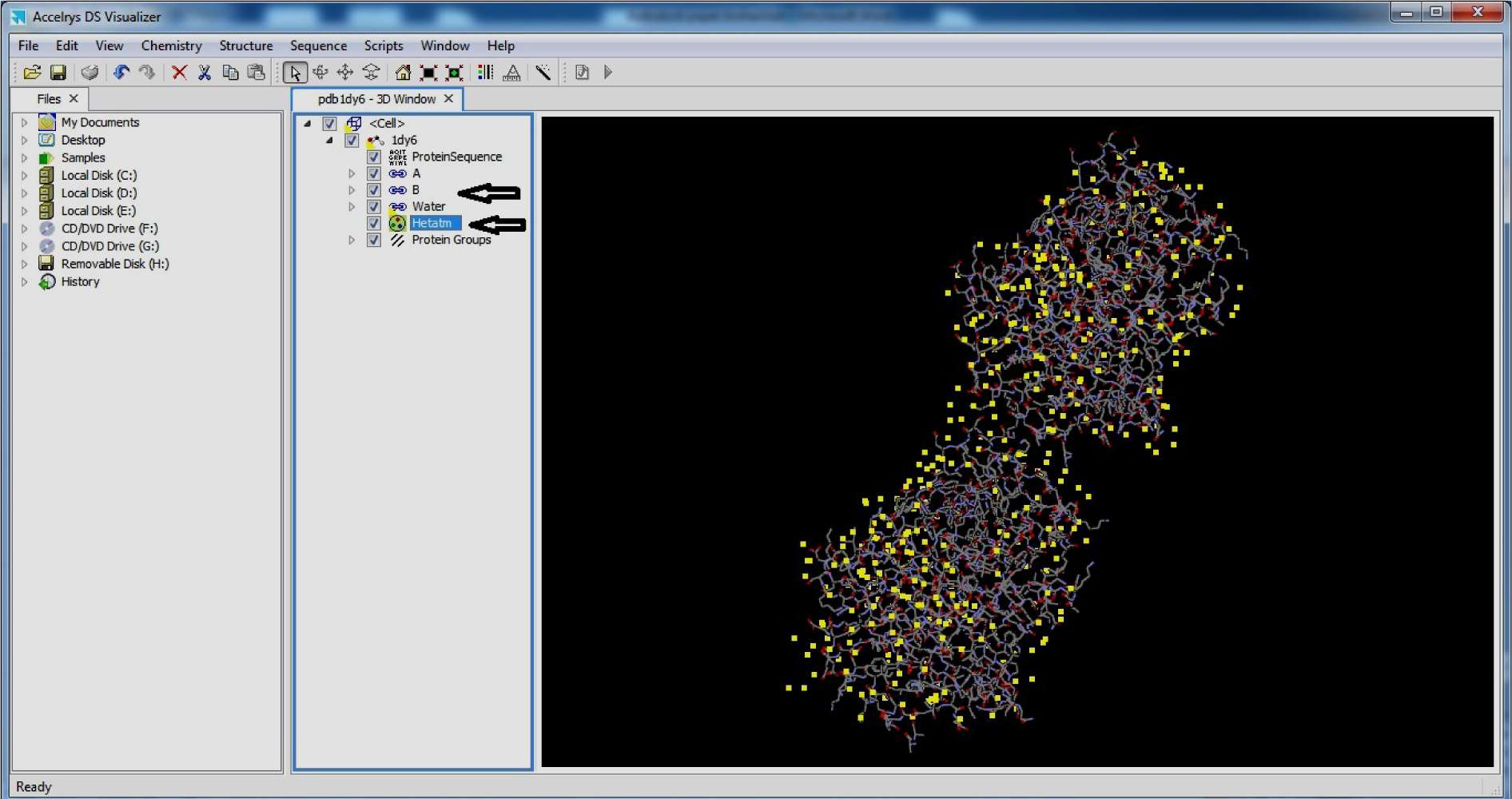
- Press Control+H
- Select Hetatm and Delete
(Fig. 7)
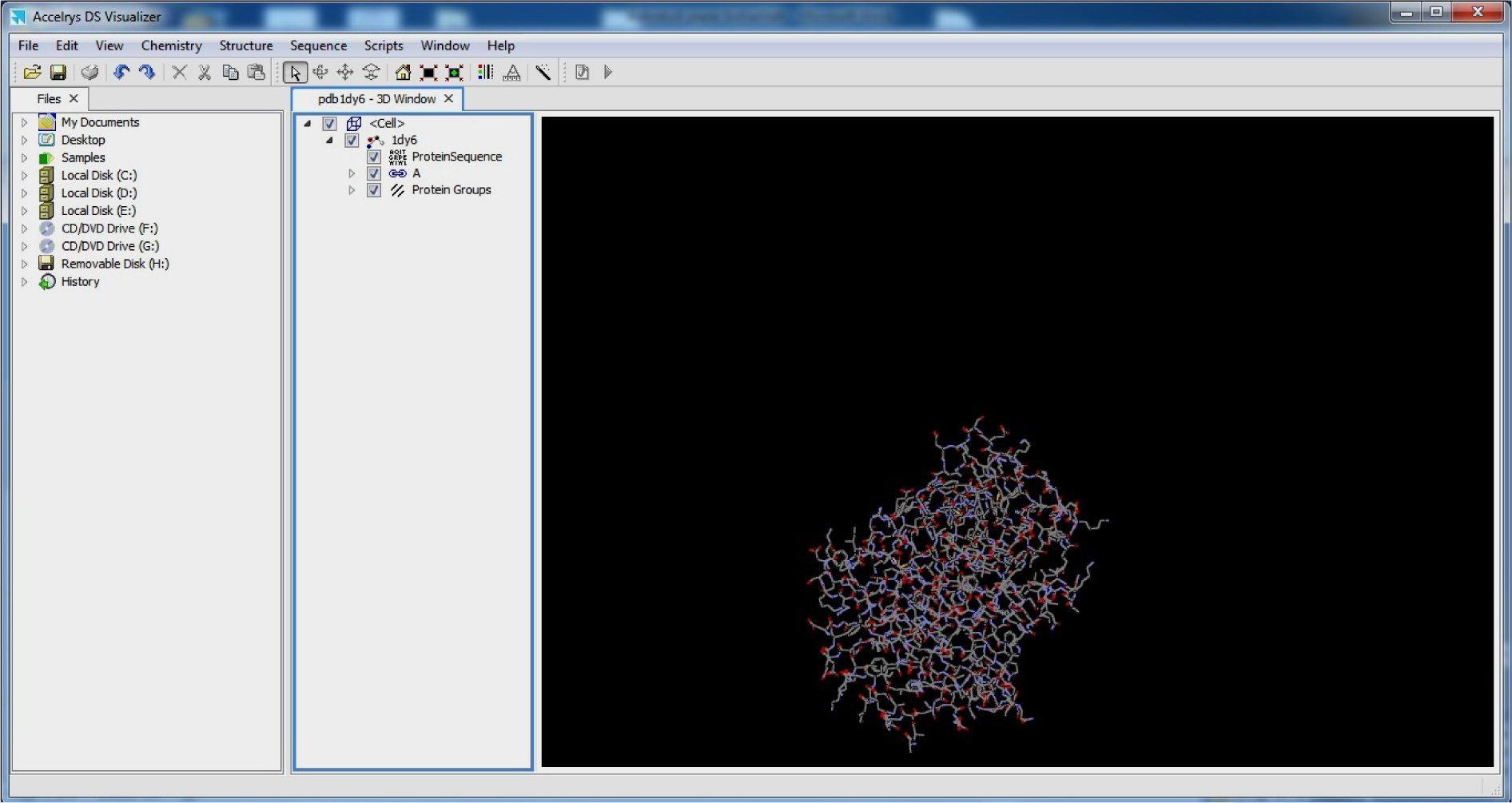
- Select B chain and Delete
(As both A and B chain are similar and Imipenemcan bind to anyone of the two chains)
(Fig. 8)
Save as Target.pdb
(Fig. 9)
1.2 Retrieving Ligand.pdb files from major ligand databases
http://pubchem.ncbi.nlm.nih.gov/
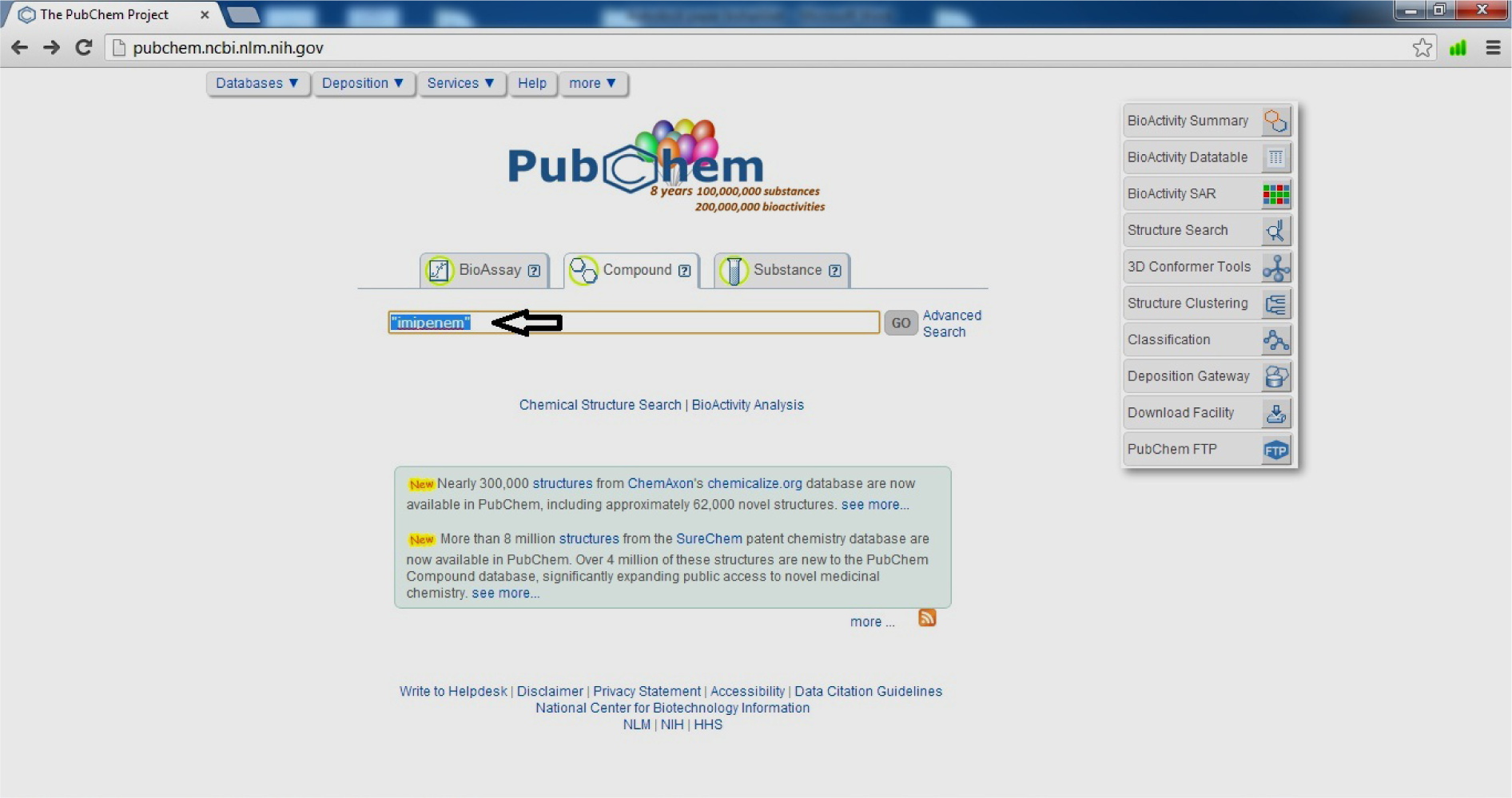
- Search your Ligand (Imipenem)
(Fig. 10)
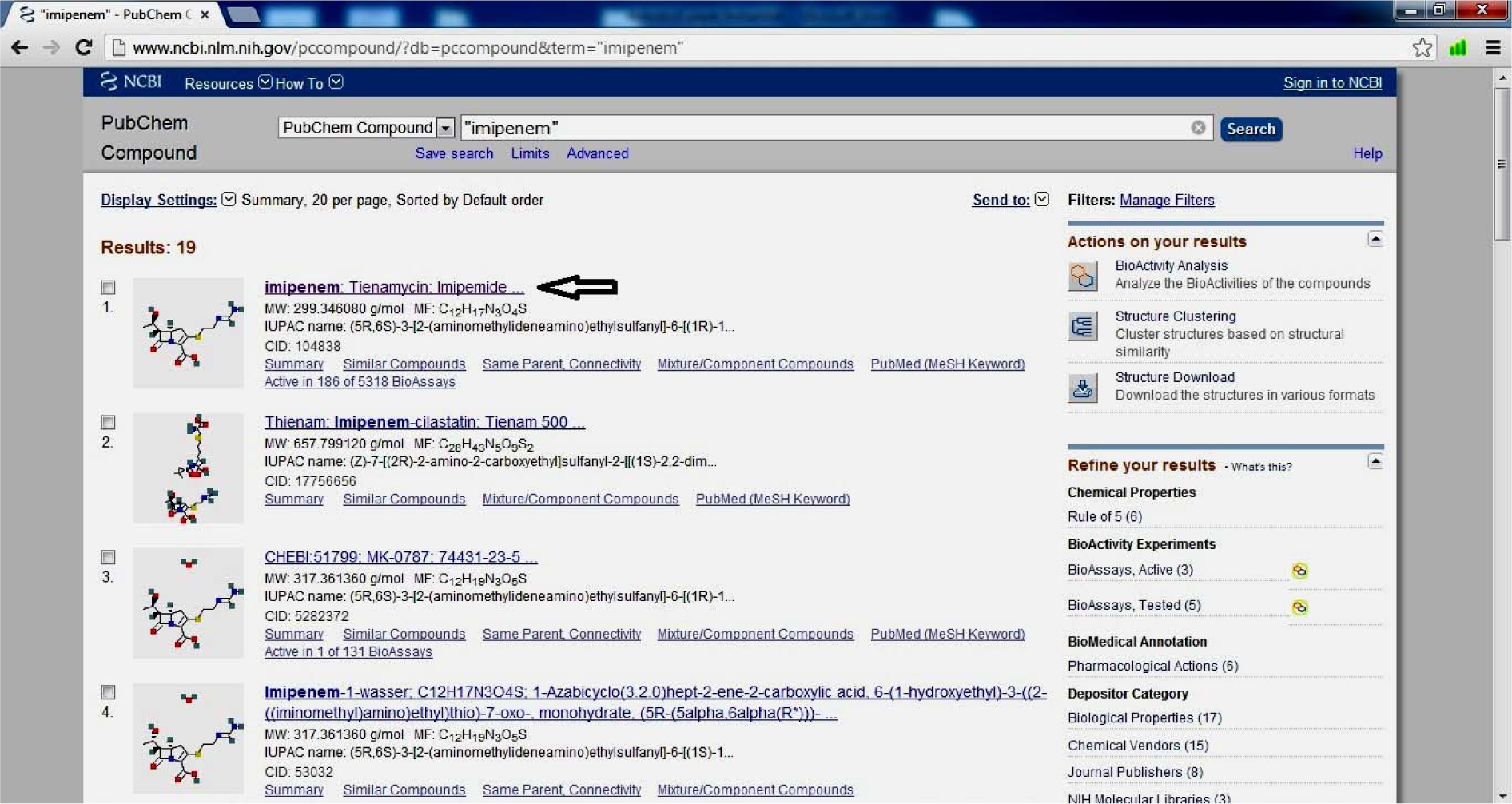
- Click on Ligand (Imipenem)
(Fig. 11)
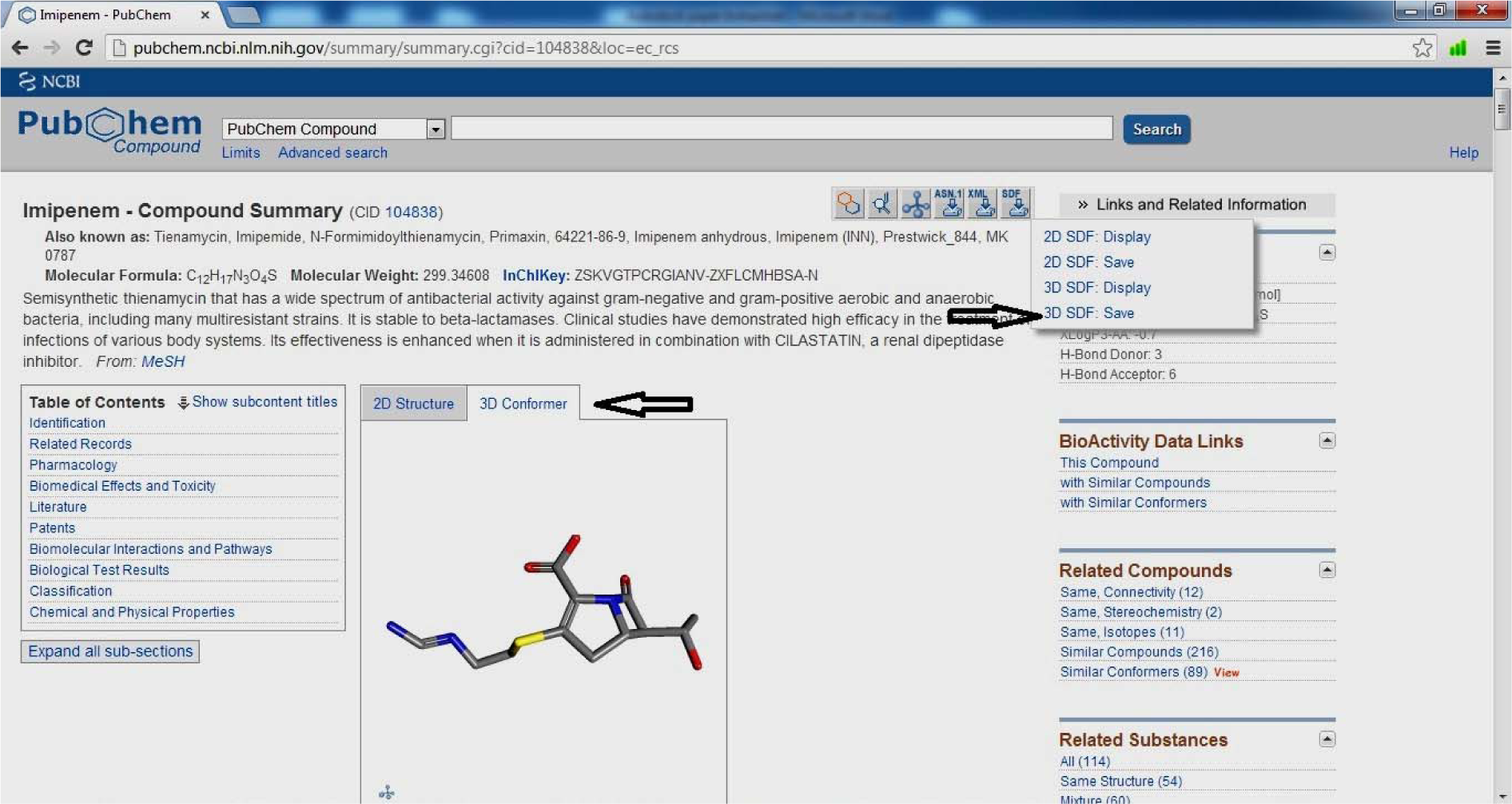
- Click 3D image
- Open SDF
- Save 3D SDF
(Fig. 12)
- Open 3D SDF file of Ligand in Discovery Studio visualiser
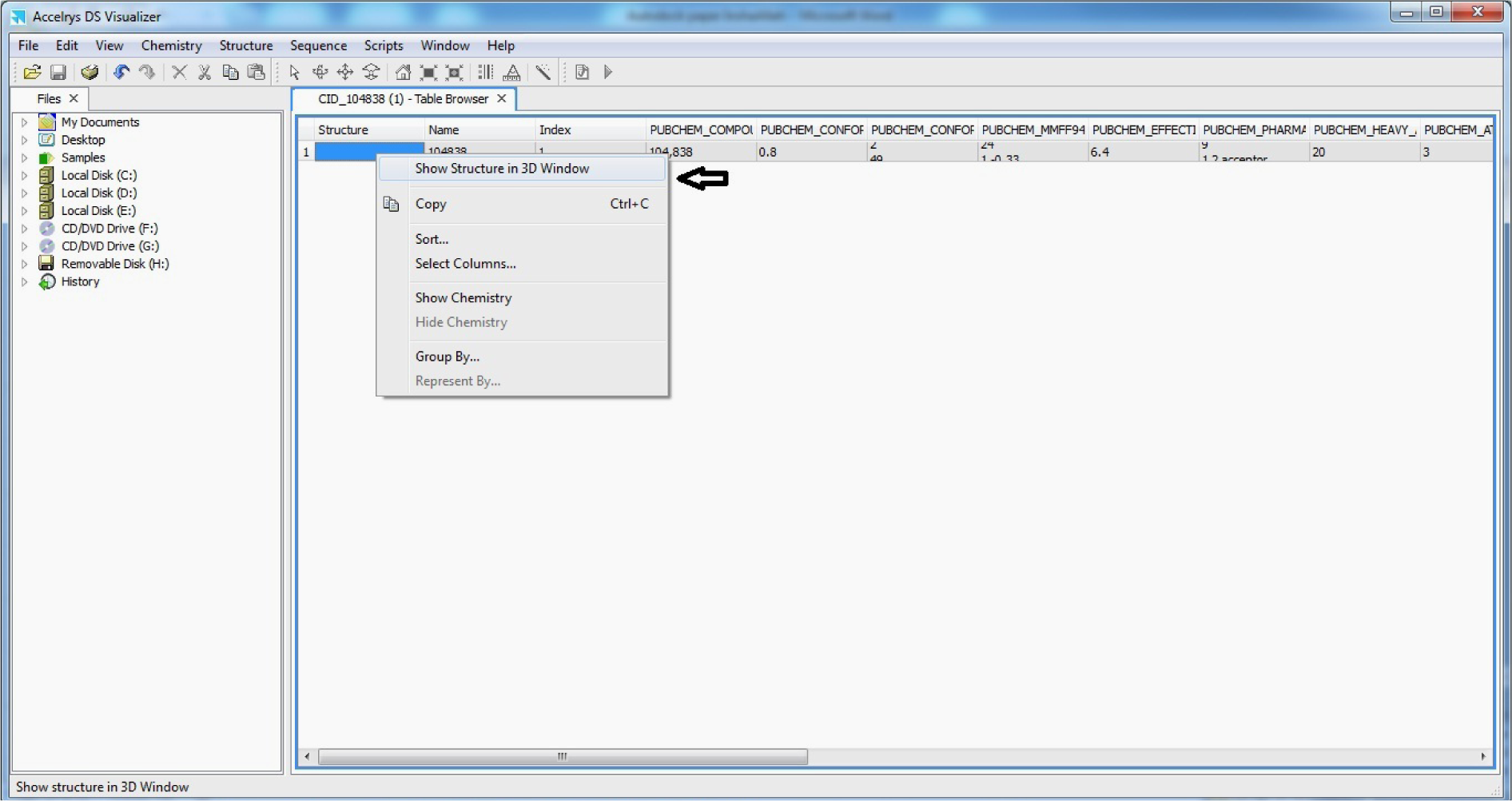
- Right Click to ‘show structure in 3D window’
(Fig. 13)
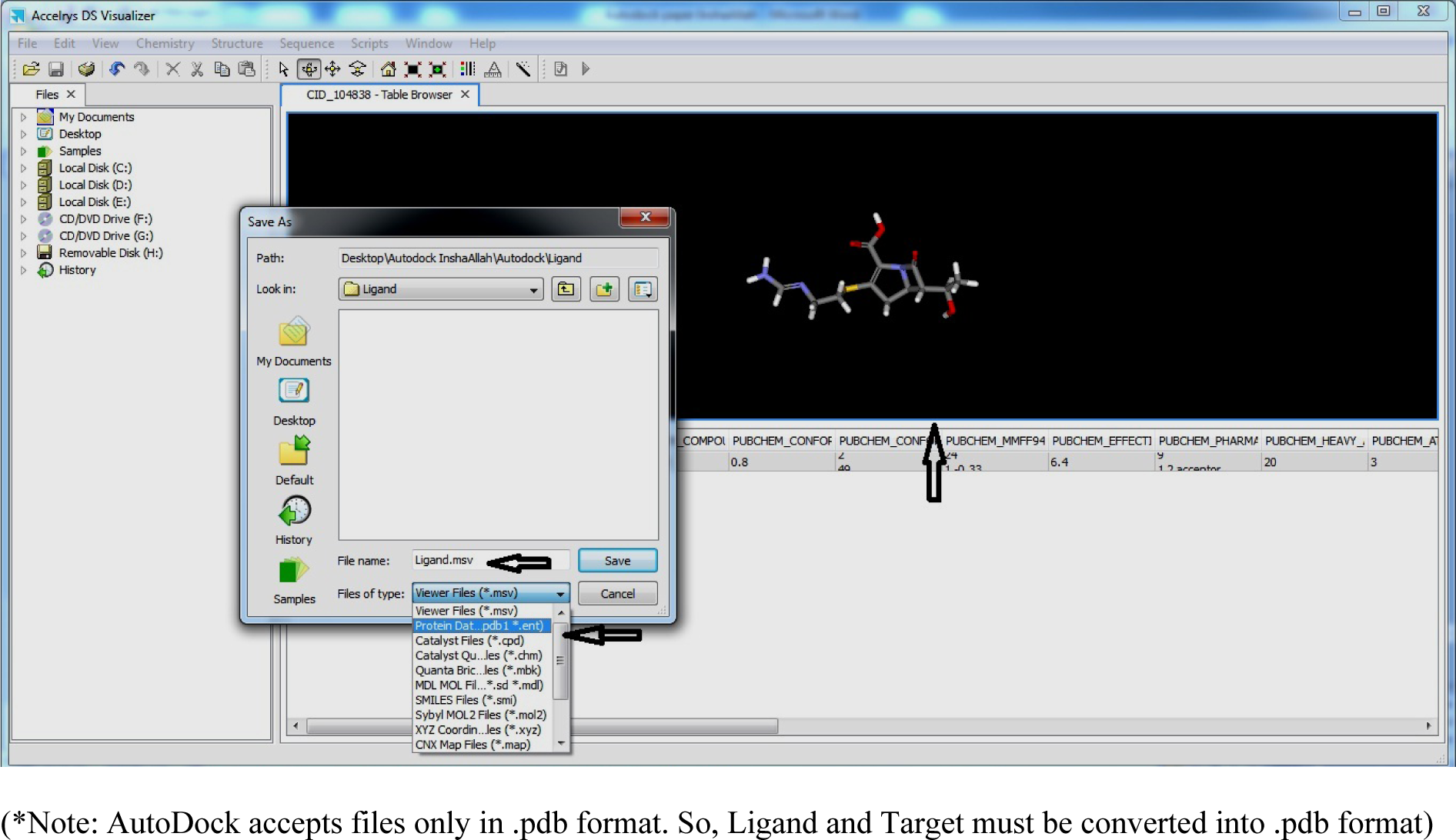
- Click on 3D image and Save as Ligand.pdb file
(Fig. 14)
2 Preparing PDBQT format for Target and ligand (Target.pdbqt, Ligand.pdbqt), Grid and Docking Parameter file (a.gpf and a.dpf) using AutoDock 4.2
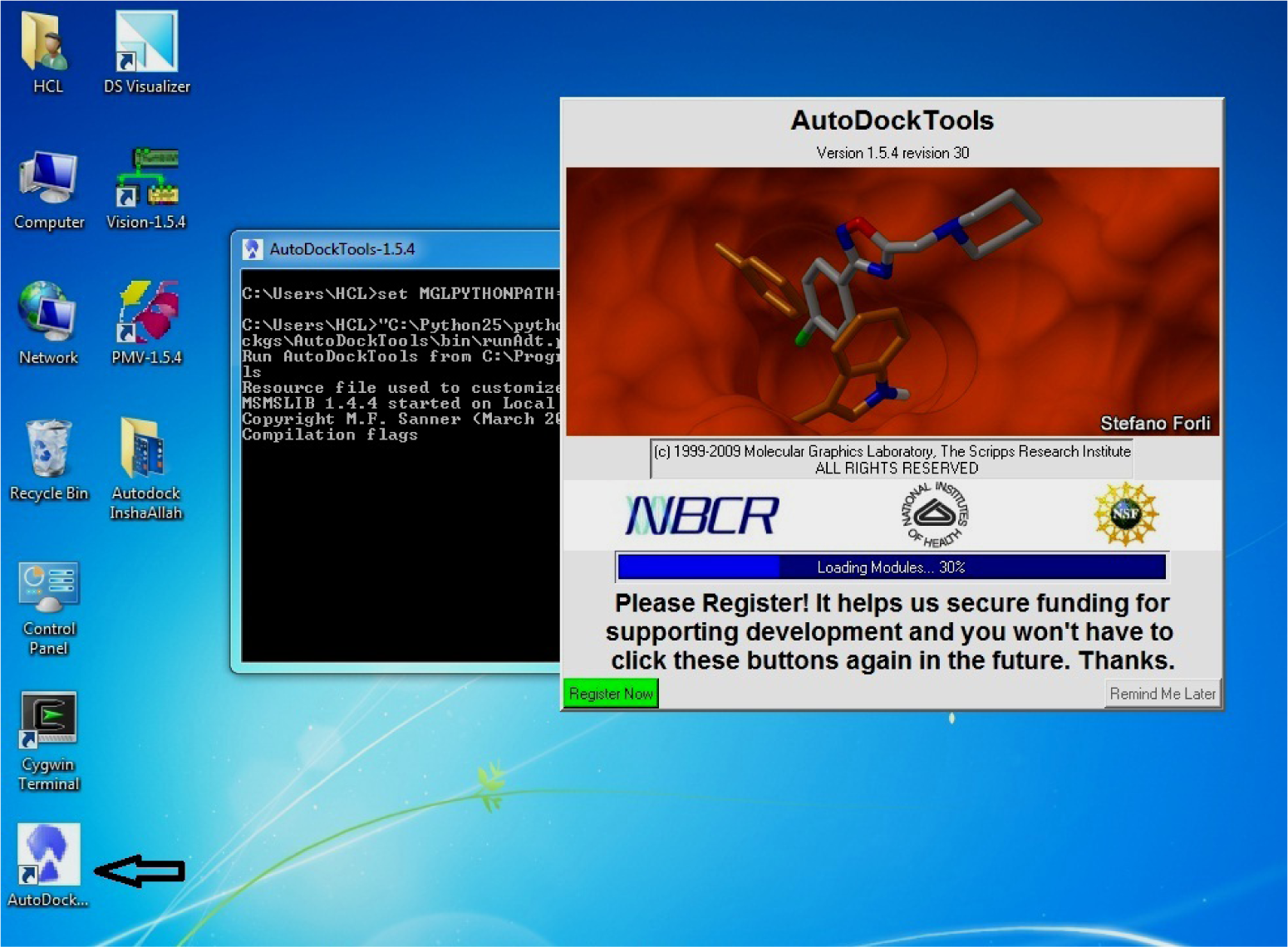
- Open AutoDock present on desktop
(*Created after successful installation of MGL Tools)
(Fig. 15)
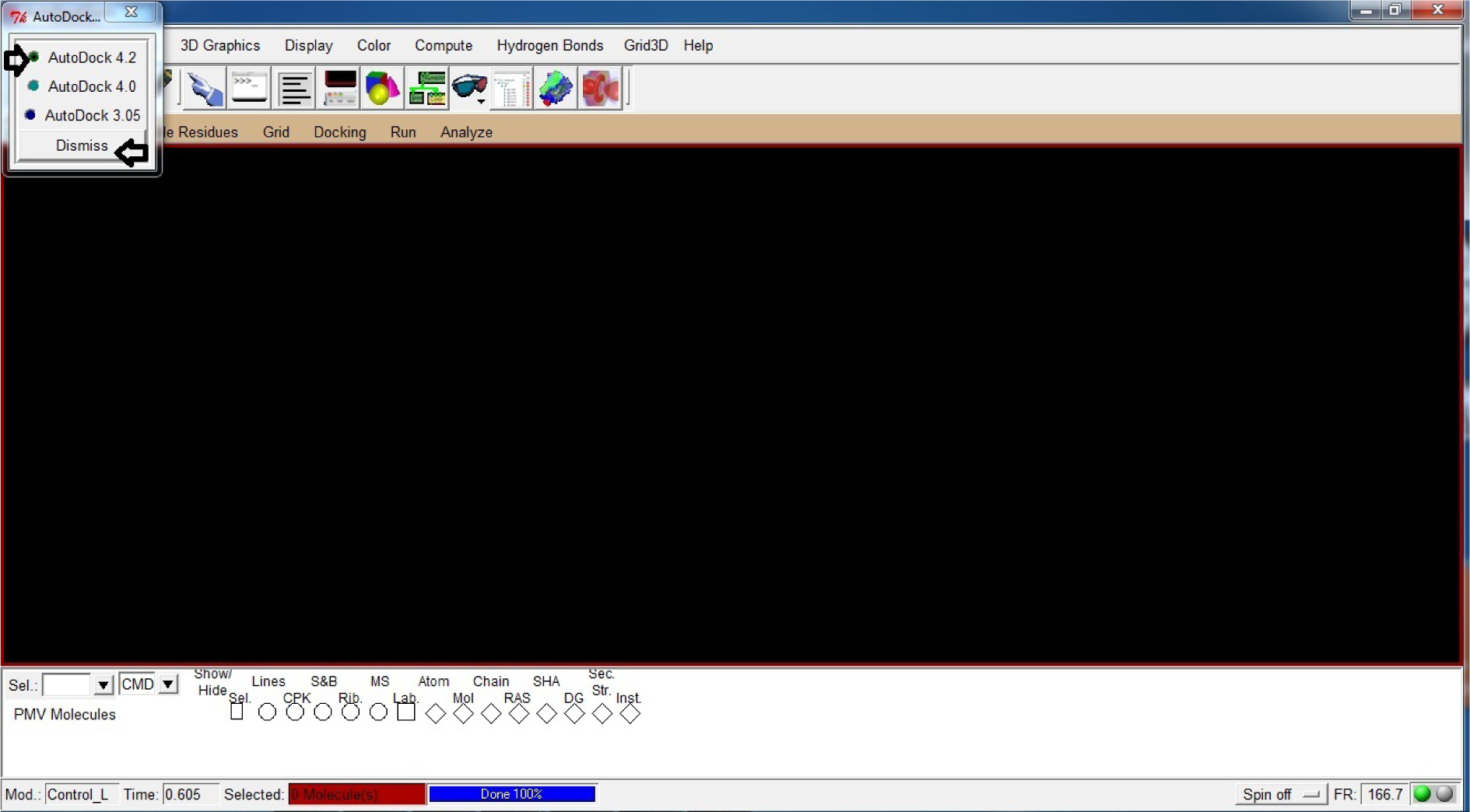
- Select AutoDock 4.2
- Dismiss
(Fig. 16)
2.1 Preparation of Target.pdbqt file
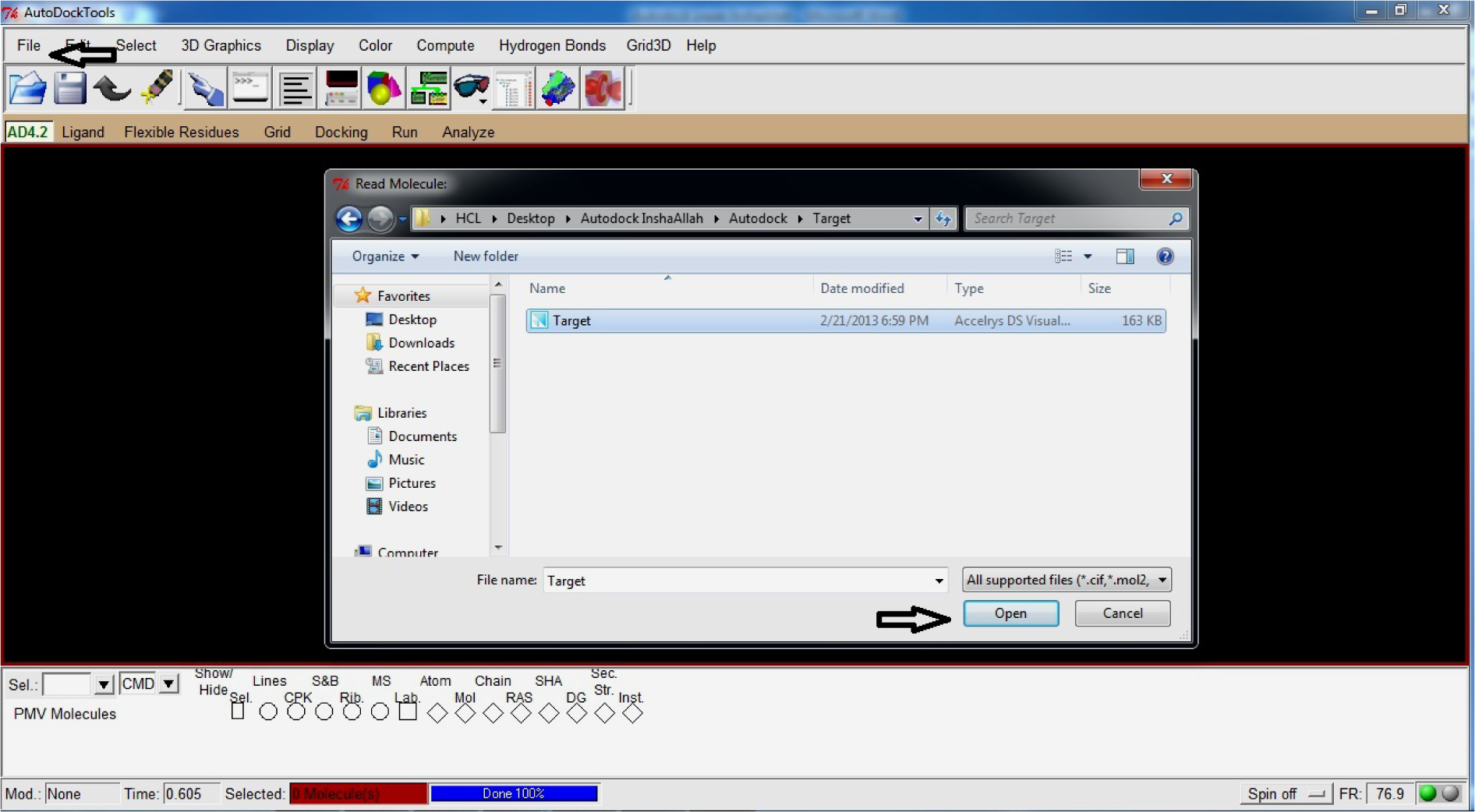
- Open File
- Read Molecule
- Select and Open Target.pdb (*Created in first step)
(Fig. 17)
- Target molecule will appear on screen
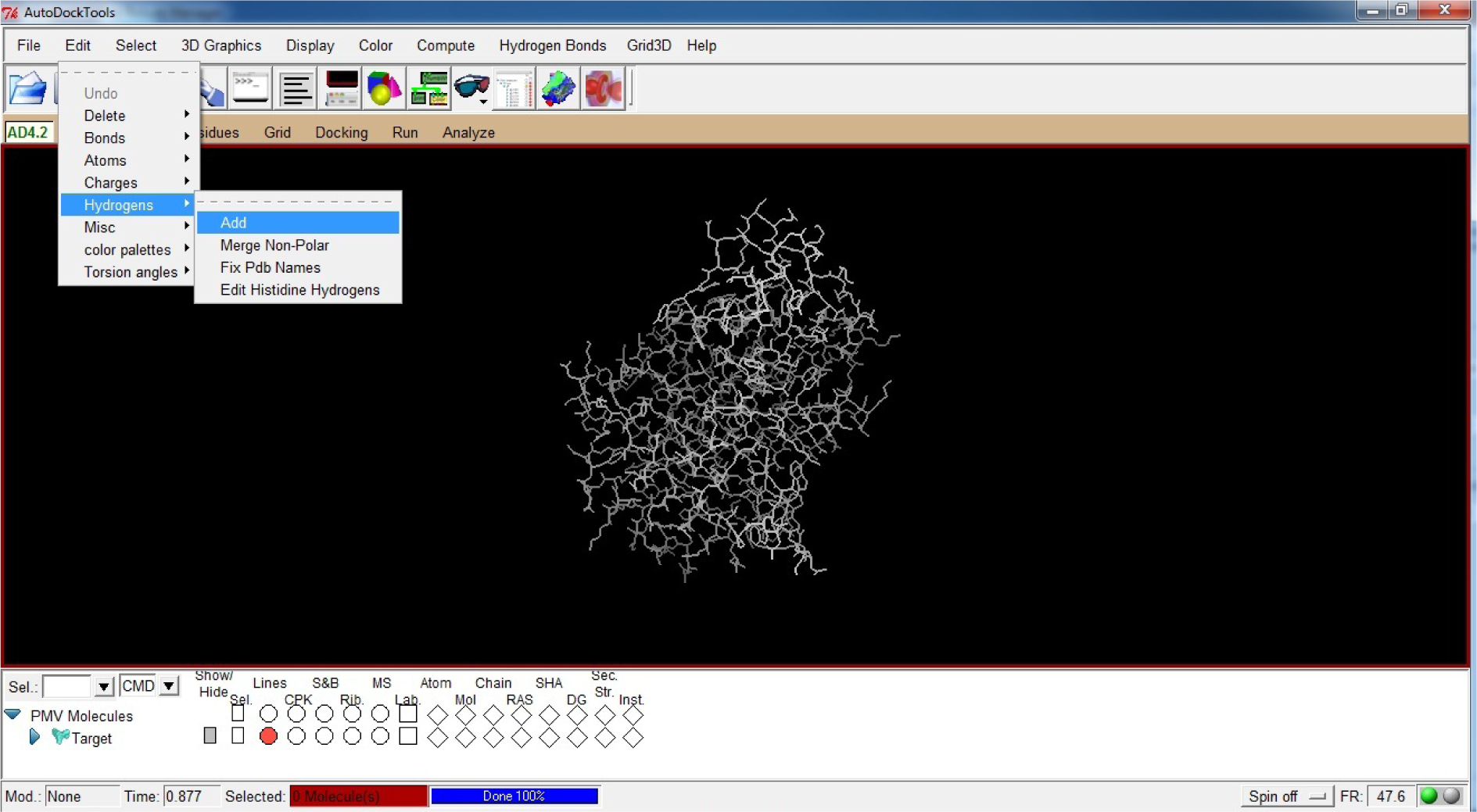
- Click on Edit
- Click on Hydrogens
- Click on Add
(Fig. 18)
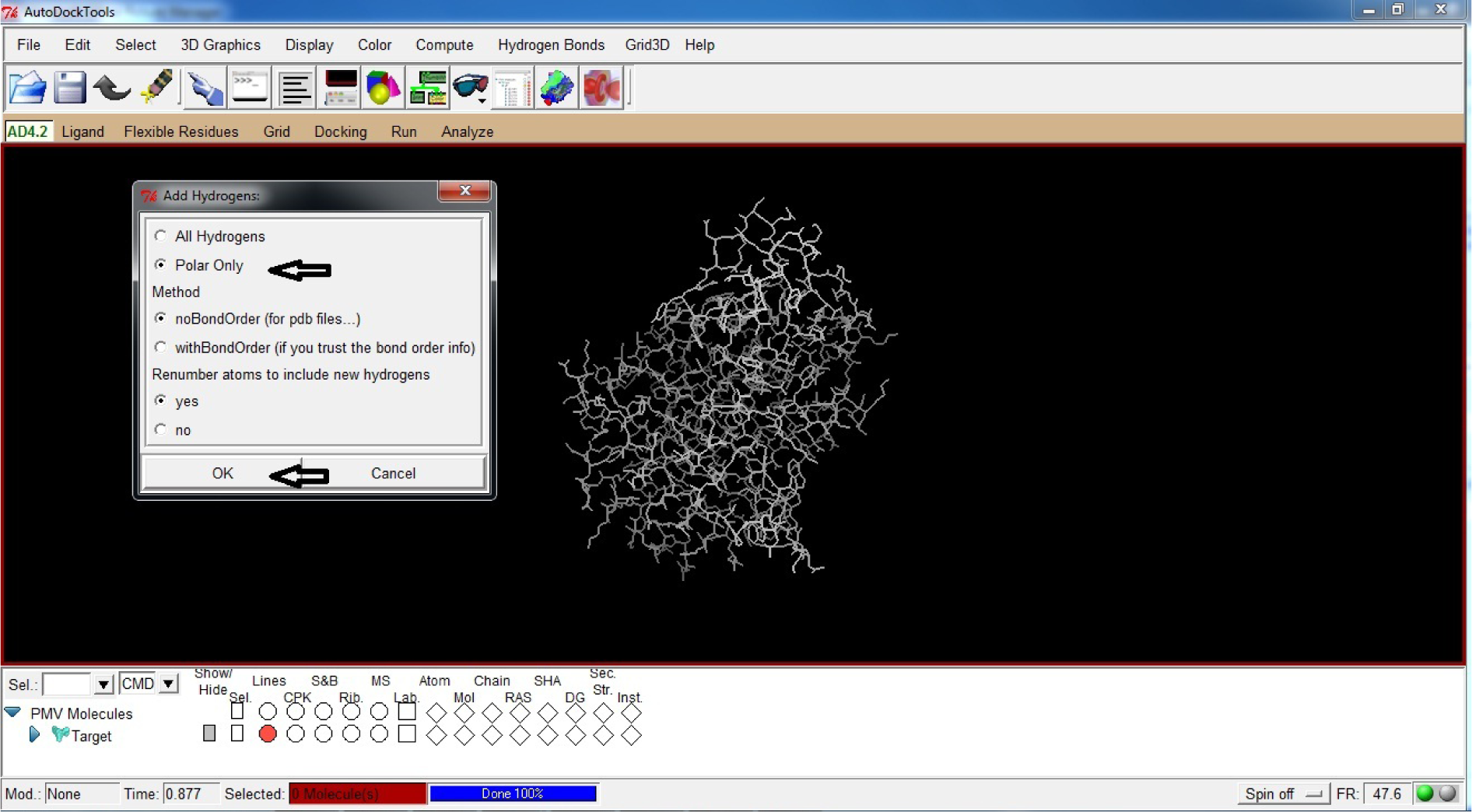
Click Polar Only
Click OK
(Fig. 19)
- Again Edit
- Click Charges
- Add Kollman Charges
- Click OK
- Open Grid
- Click on Macromolecules
- Click on Choose
- Click Target
- Click Select Molecule
- Click OK
(Fig. 20)
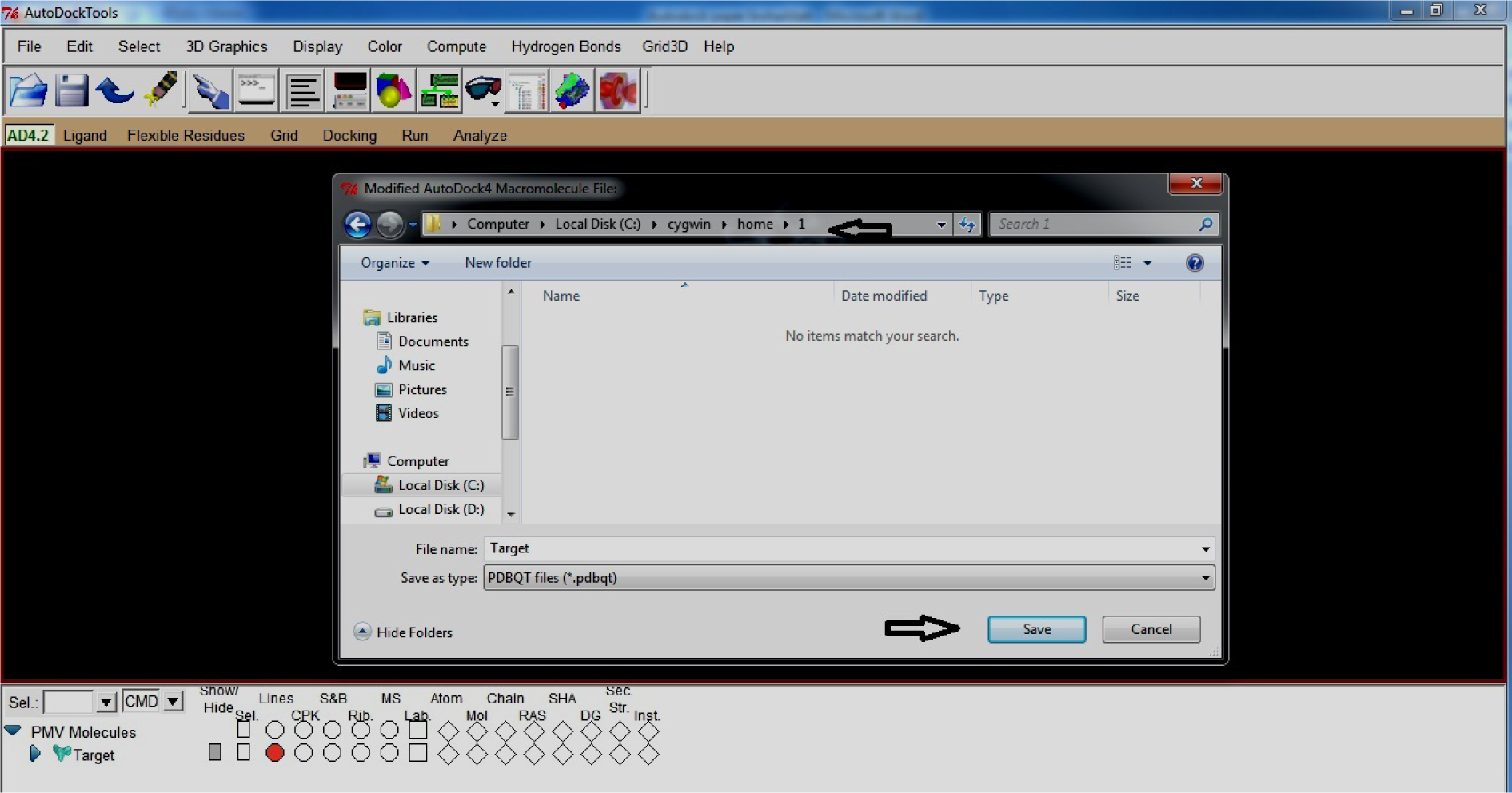
- Open My computer
- Open C drive
- Open Cygwin
- Open home
- Create new folder and rename it as 1 (or any other shortname)
- Save Target in Folder 1
(*In short: save Target.pdbqt in C:\Cygwin\home\1 and after saving macromolecule gets coloured)
(Fig. 21)
2.2 Preparation of Ligand.pdbqt file
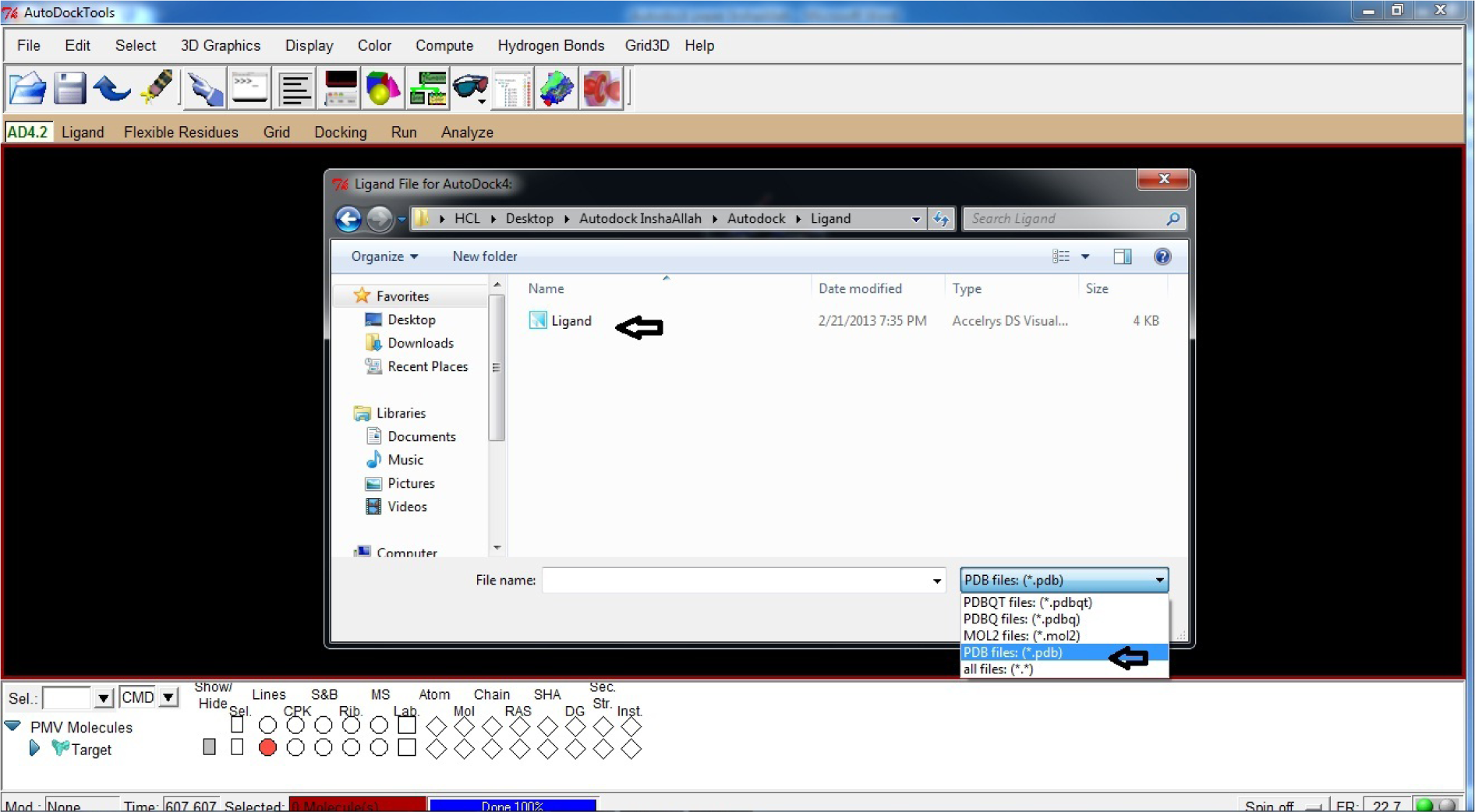
- Open Ligand
- Click Input
- Click Open
- Change format from .pdbqt to .pdb
(Fig. 22)
- Select Ligand
- Click Open
- Click OK
- Again Open Ligand
- Click Torsion Tree
- Click Detect Root
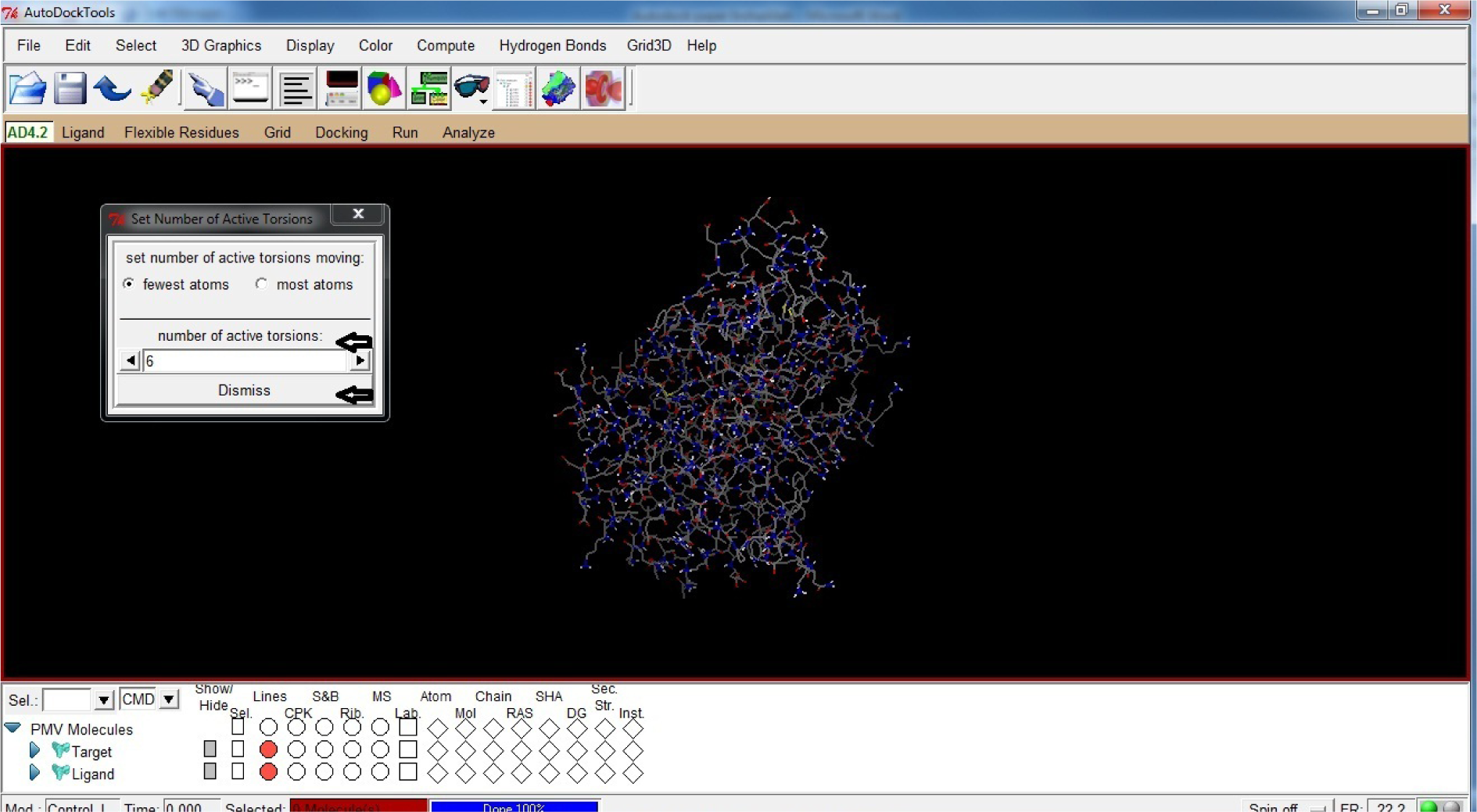
- Again Open Ligand
- Click Torsion Tree
- Click Set Number of Torsions
- Set number of active torsions between 1 to 6
- Click Dismiss
(Fig. 23)
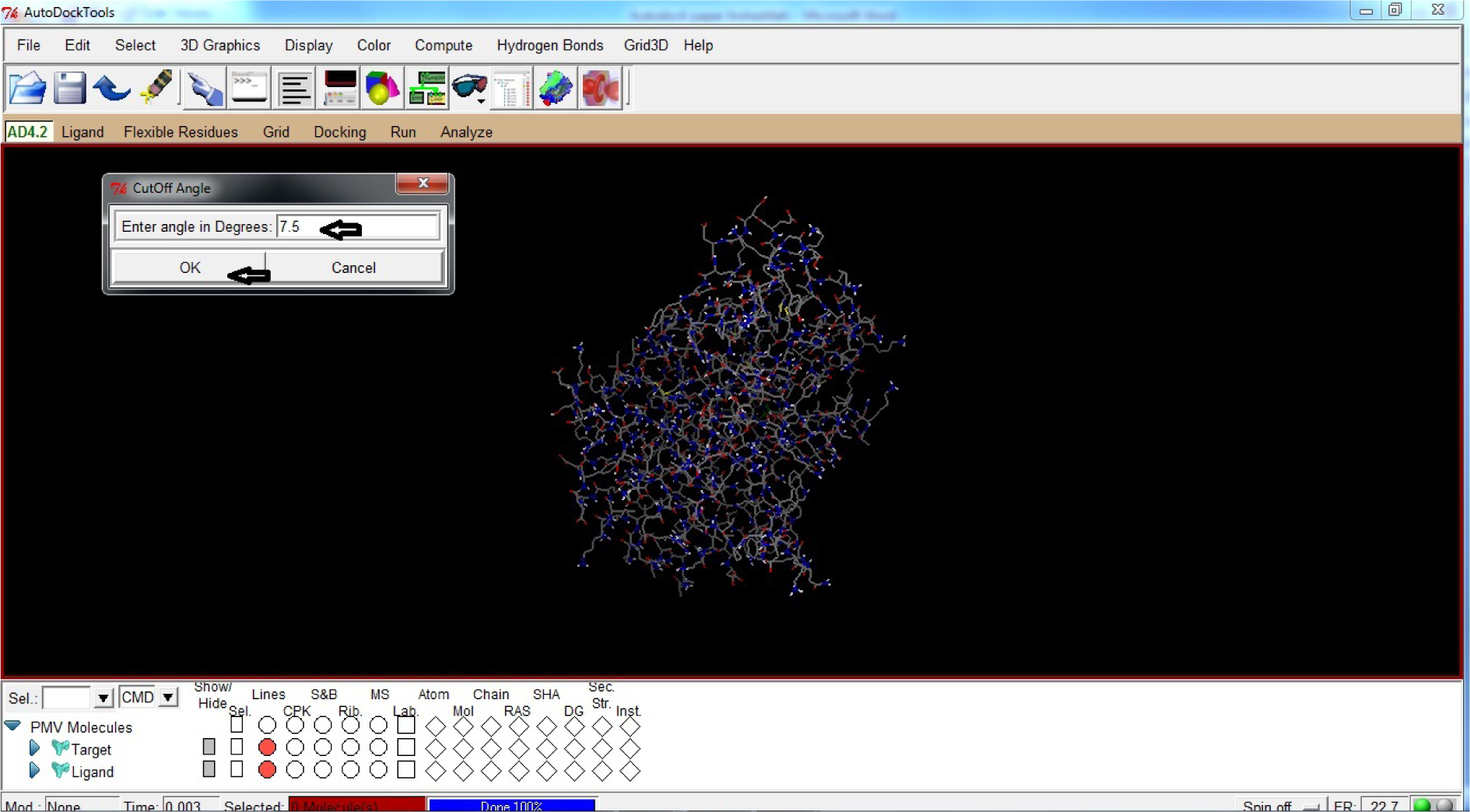
- Again Open Ligand
- Click Aromatic Carbons
- Click Aromaticity criterion
- Click OK (* If ‘Enter angle in Degrees: 7.5’)
(Fig. 24)
- Again Open Ligand
- Click Output
- Click Save as PDBQT
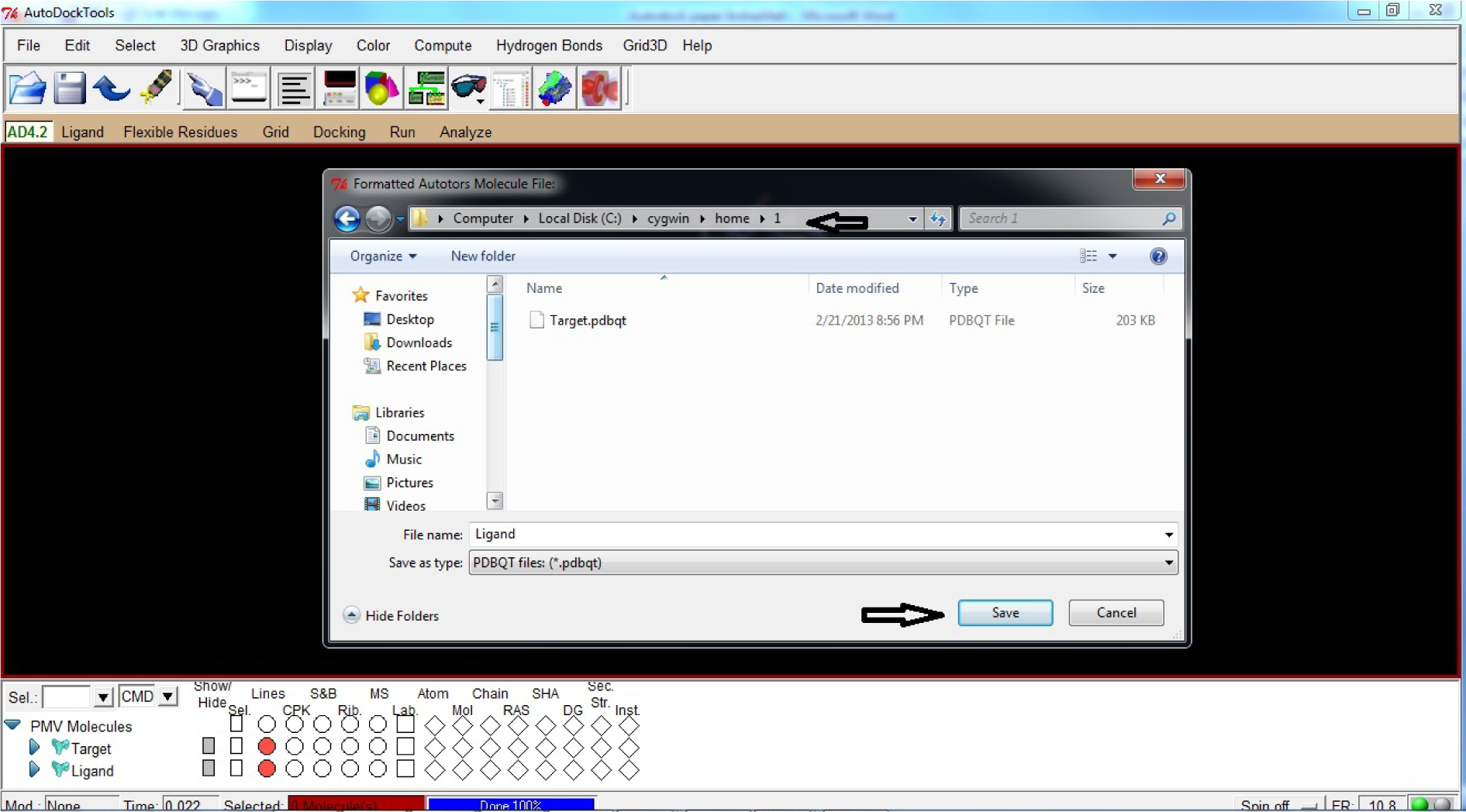
- Save Ligand file in C:\Cygwin\home\1
(* In the same folder and in same way as Target.pdbqt file)
(Fig. 25)
2.3 Preparation of Grid Parameter File (a.gpf)
- Open Grid
- Click Set Map Types
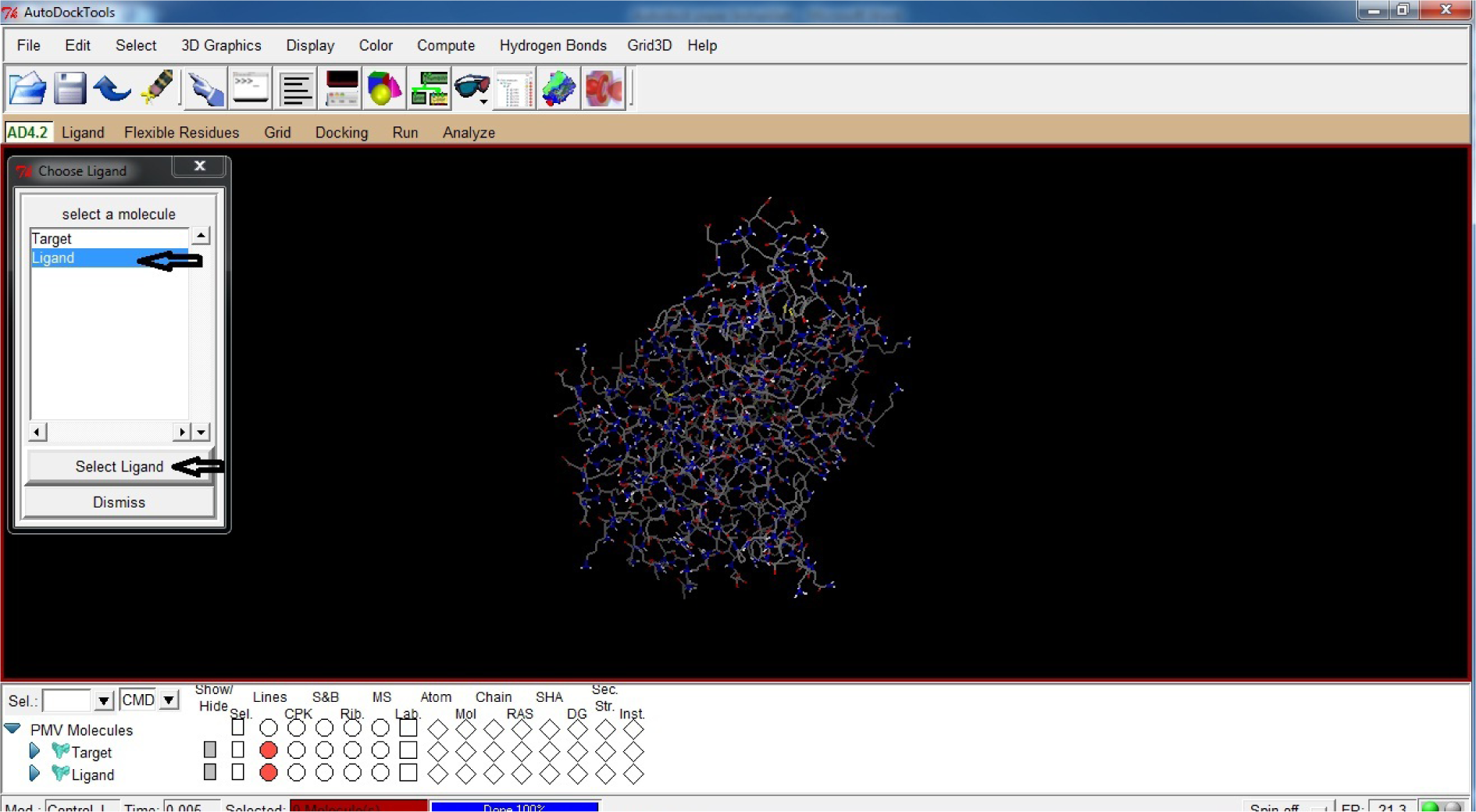
- Click Choose Ligand
- Click Ligand
- Click Select Ligand
(Fig. 26)
- Again Open Grid
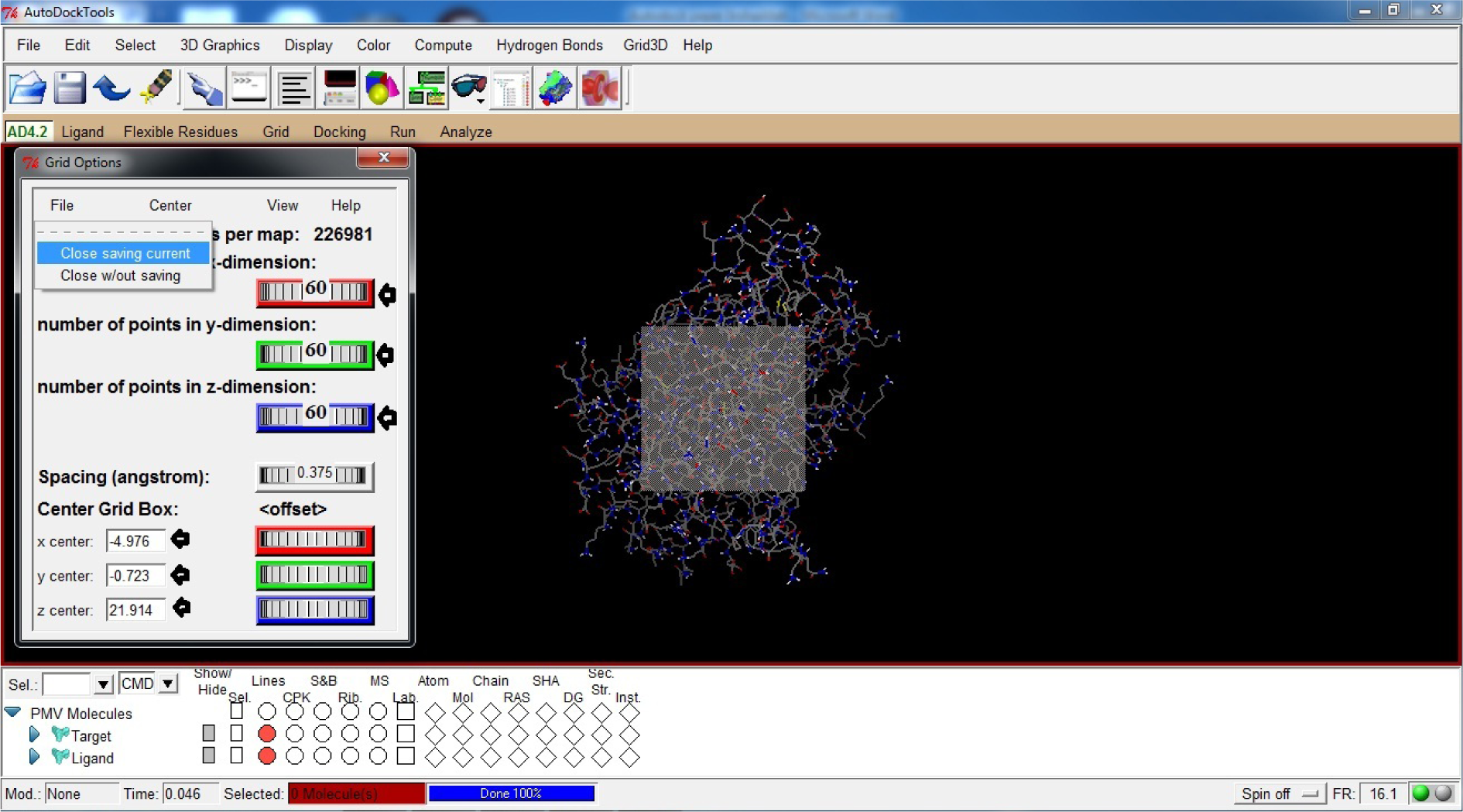
- Click Grid Box
(*We have used X,Y,Z dimension as 60x60x60. Further X,Y,Zcenter (Center Grid Box) can be changed according to the requirements but we are taking them as Default)
- Click File
- Click Close saving current
(Fig. 27)
- Again Open Grid
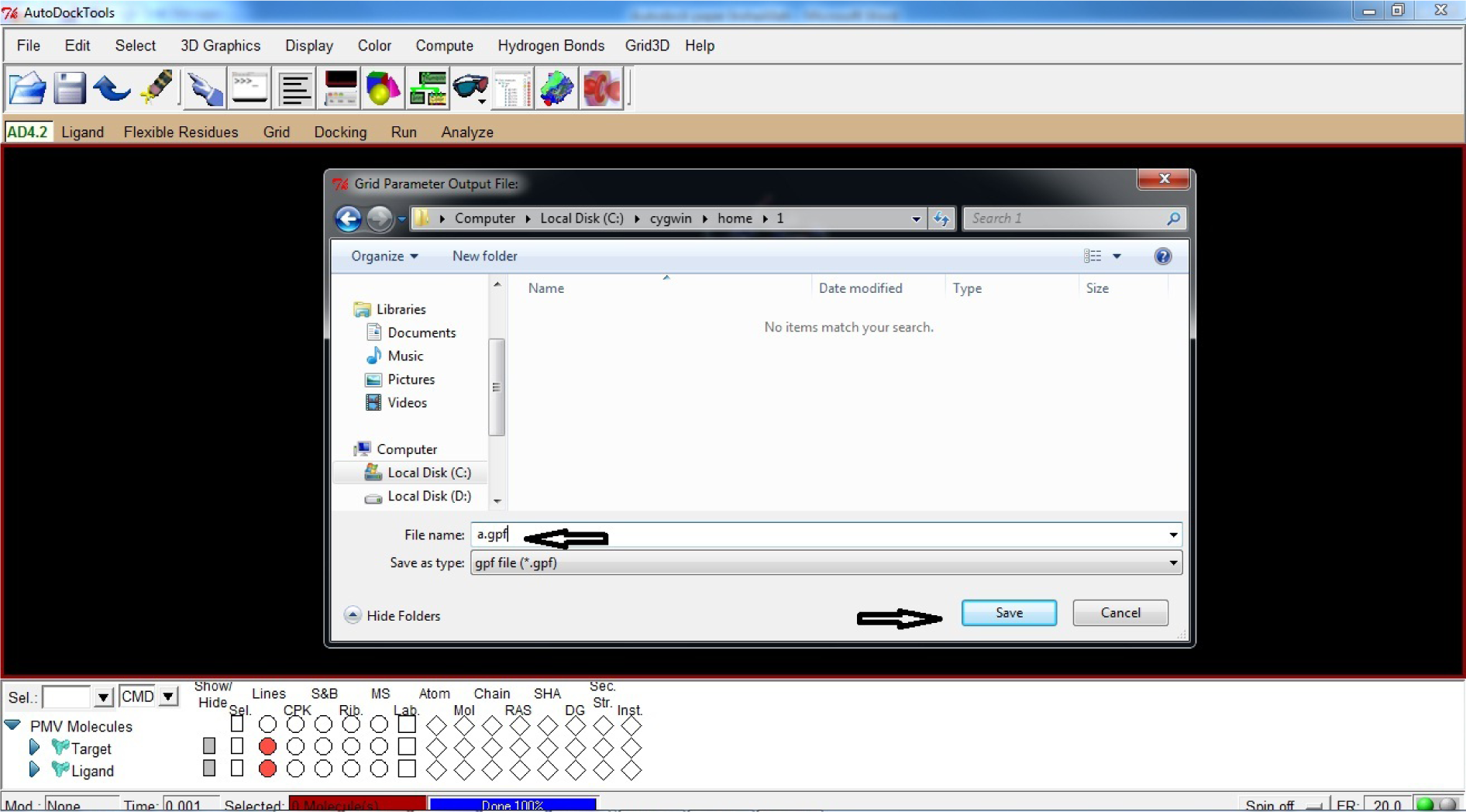
- Click Output
- Click Save GPF
- Name the File name as a.gpf
- Save a.gpf file (.gpf format) in C:\Cygwin\home\1 (* In the same file where Target and Ligand .pdbqt files were saved)
(Fig. 28)
2.4 Preparation of Docking Parameter File (a.dpf)
- Open Docking
- Click Macromolecules
- Click Set Rigid Filename
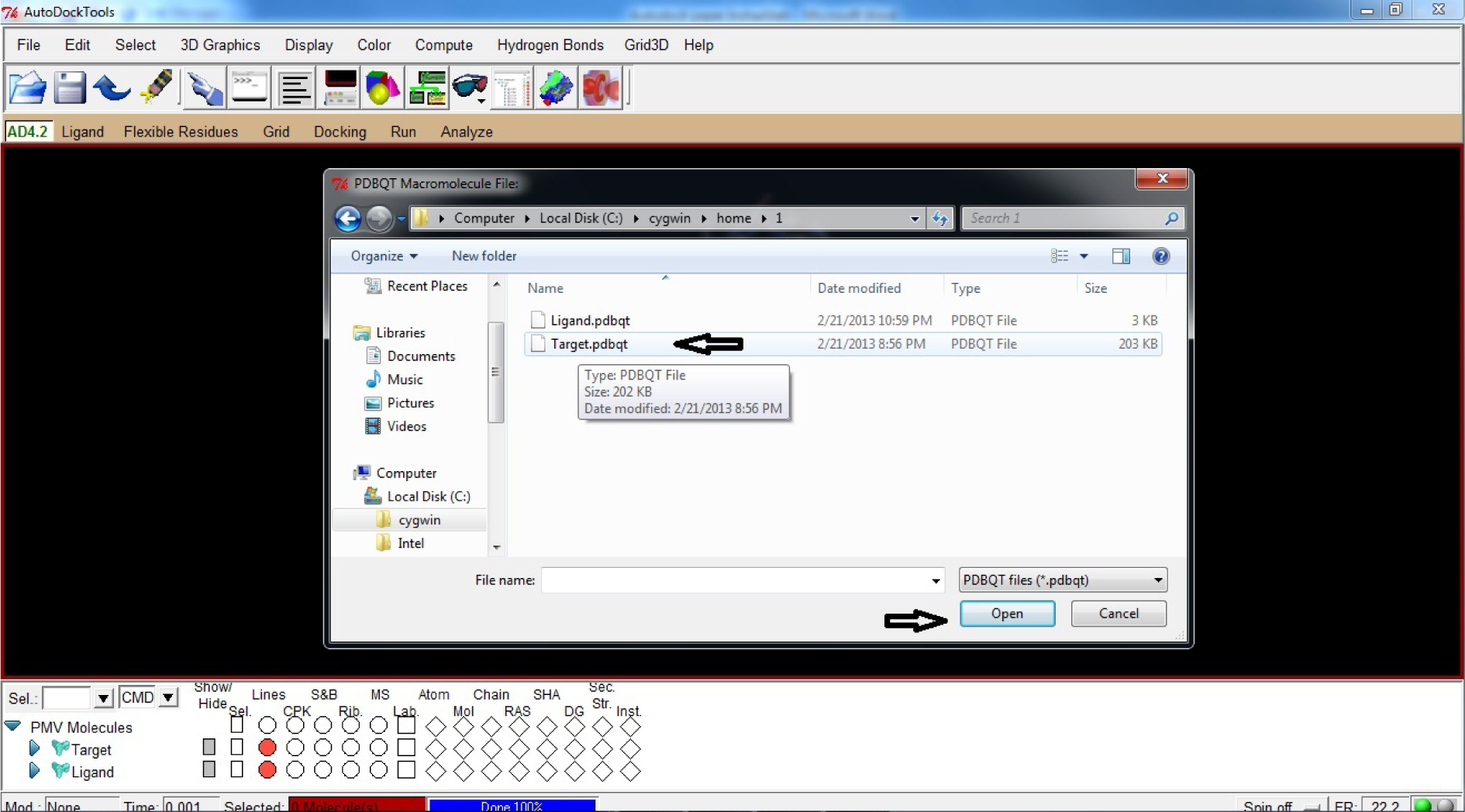
- Go to C:\ Cygwin\ home\ 1
- Select Target.pdbqt
- Click Open
(Fig. 29)
- Again Docking
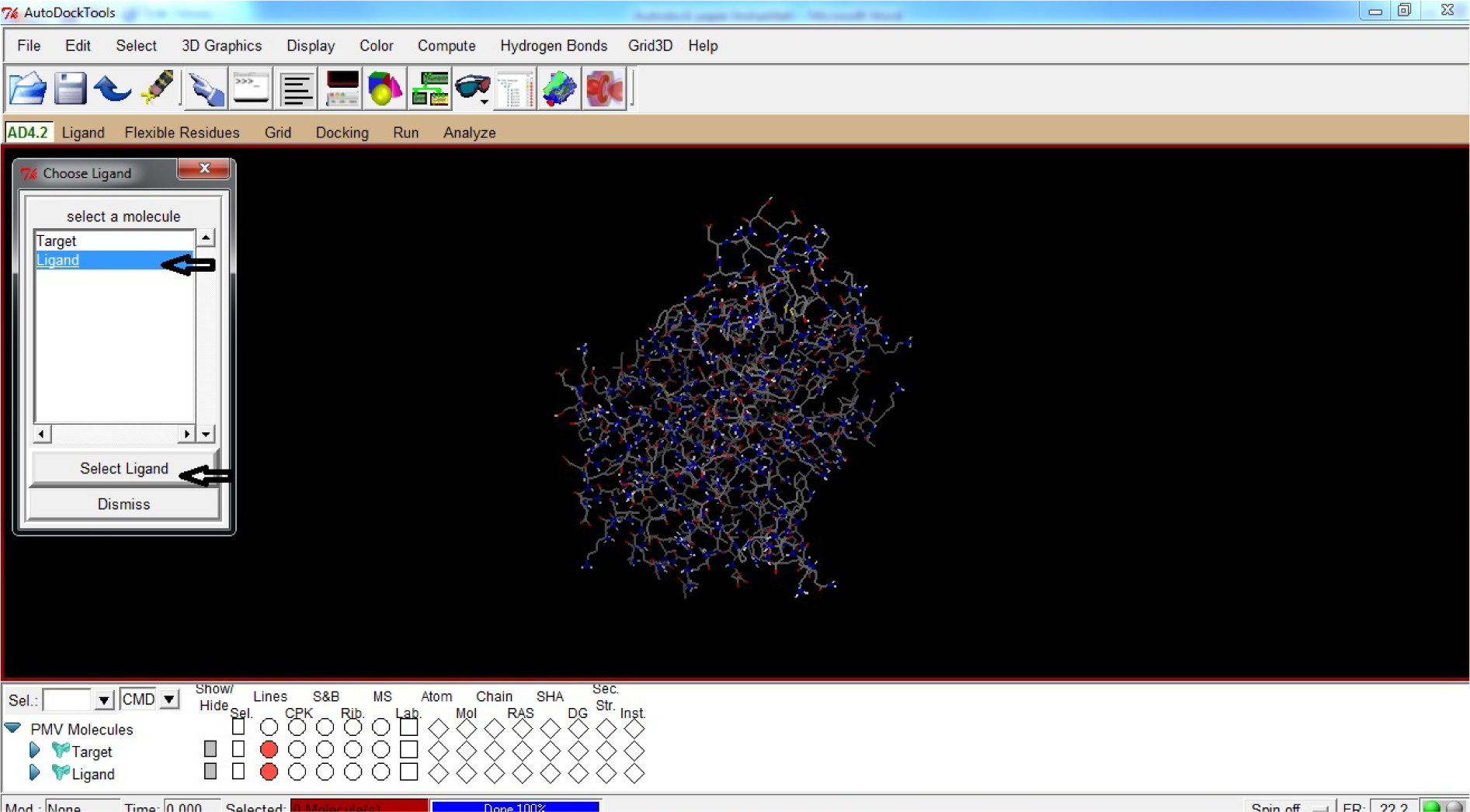
- Click Ligand
- Click Choose
- Click Ligand
- Click Select Ligand
(Fig. 30)
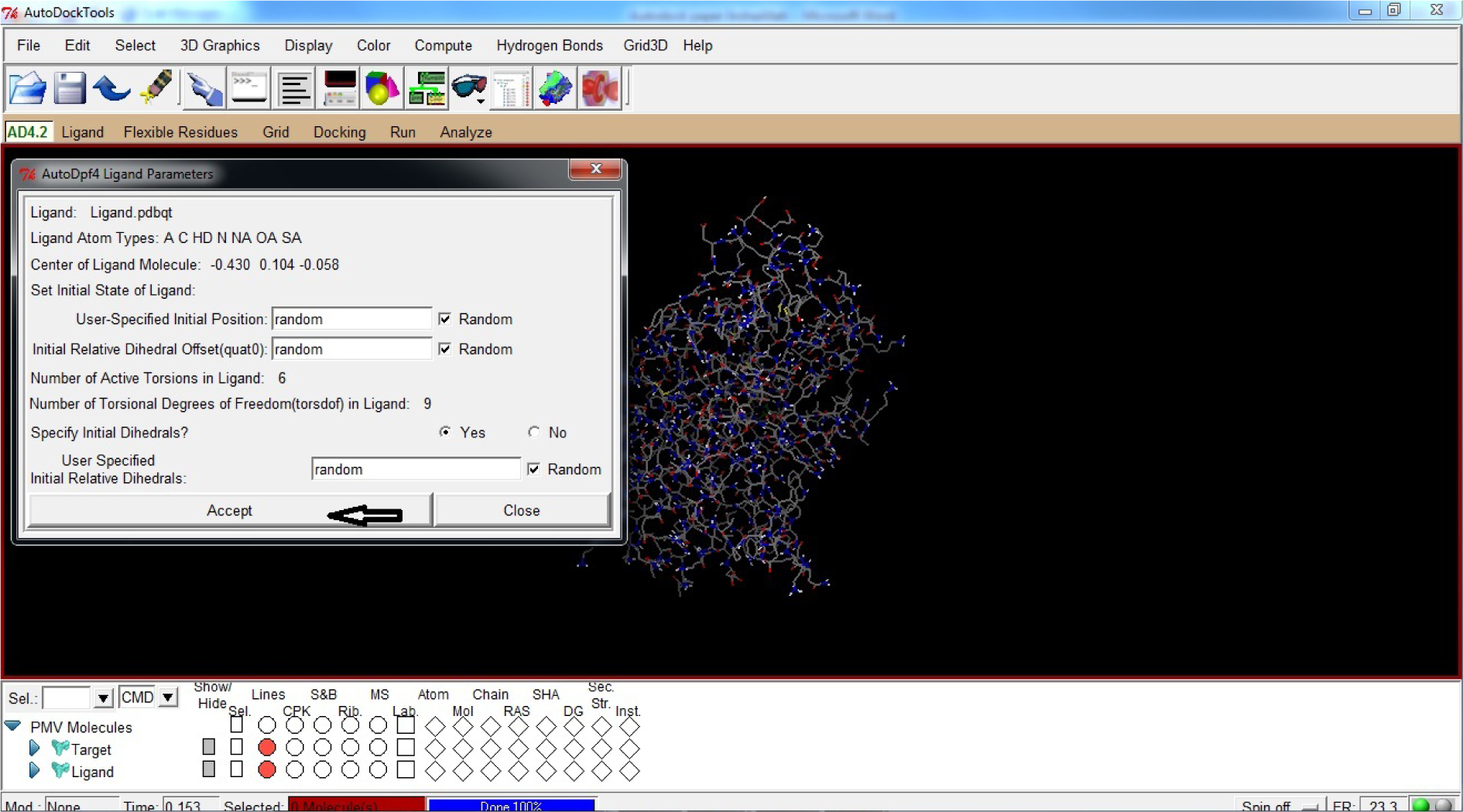
- Click Accept
(Fig. 31)
- Again Docking
- Click Search Parameters
- Click Genetic Algorithm
- Click Accept (*Using Default but we can change no. of GA runs)
- Again Docking
- Click Docking parameters
- Click Accept (*Using Default)
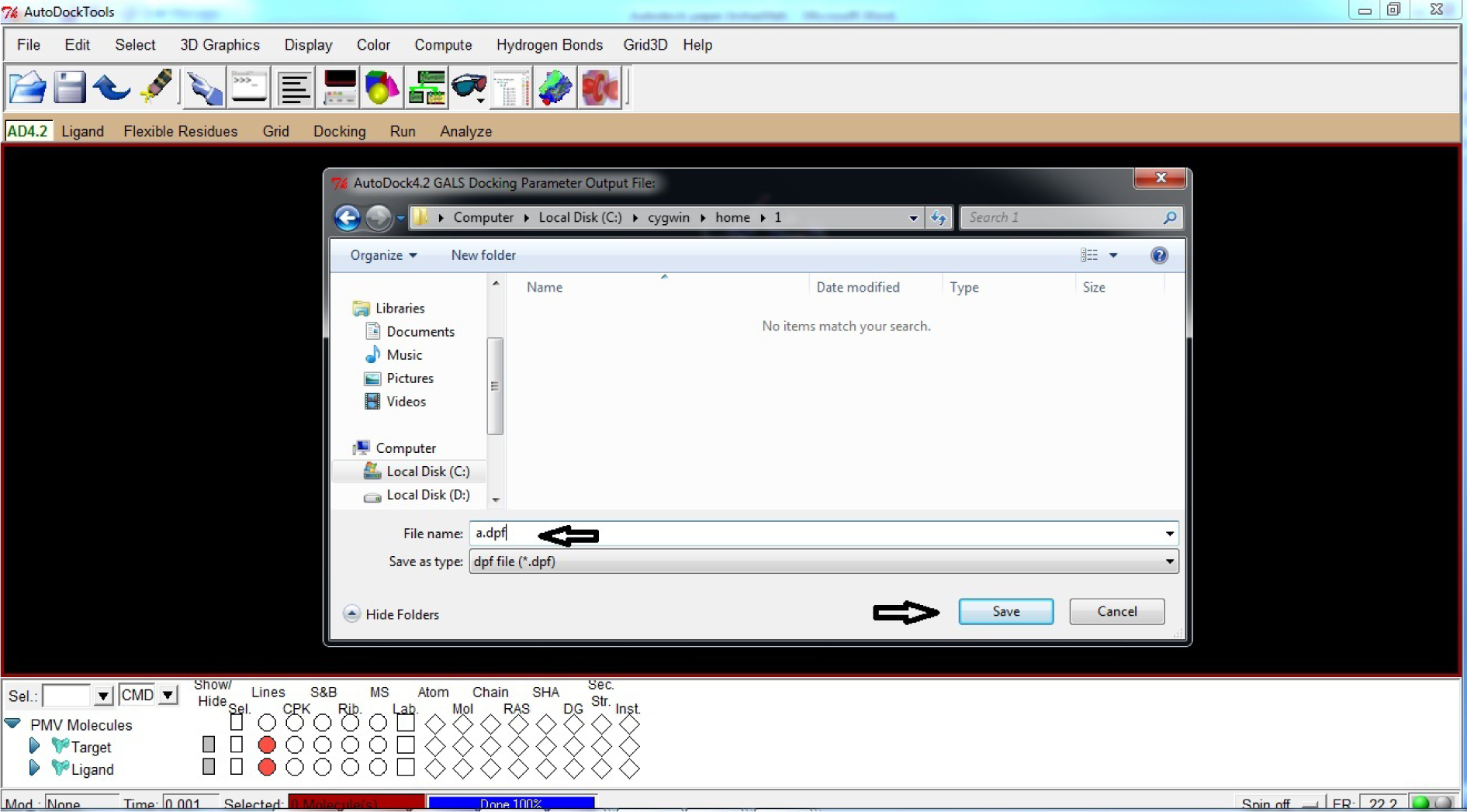
- Again docking
- Click Output
- Click LamarkianGA(4.2)
- Name the File name as a.dpf
- Save a.dpf file (.dpf format) in C:\Cygwin\home\1
(* In the same file where Target and Ligand .pdbqtand a.gpffiles were saved)
(Fig. 32)
At last four files Target.pdbqt, Ligand.pdbqt, a.gpf and a.dpf are present in the C:\ Cygwin\ home\1
(Fig. 33)
3 Using Cygwin for Molecular Docking
Open Cygwin (*By clicking icon on the desktop)
Use these commands highlighted in brown font color by copy and paste in Cygwin and press enter after each command:
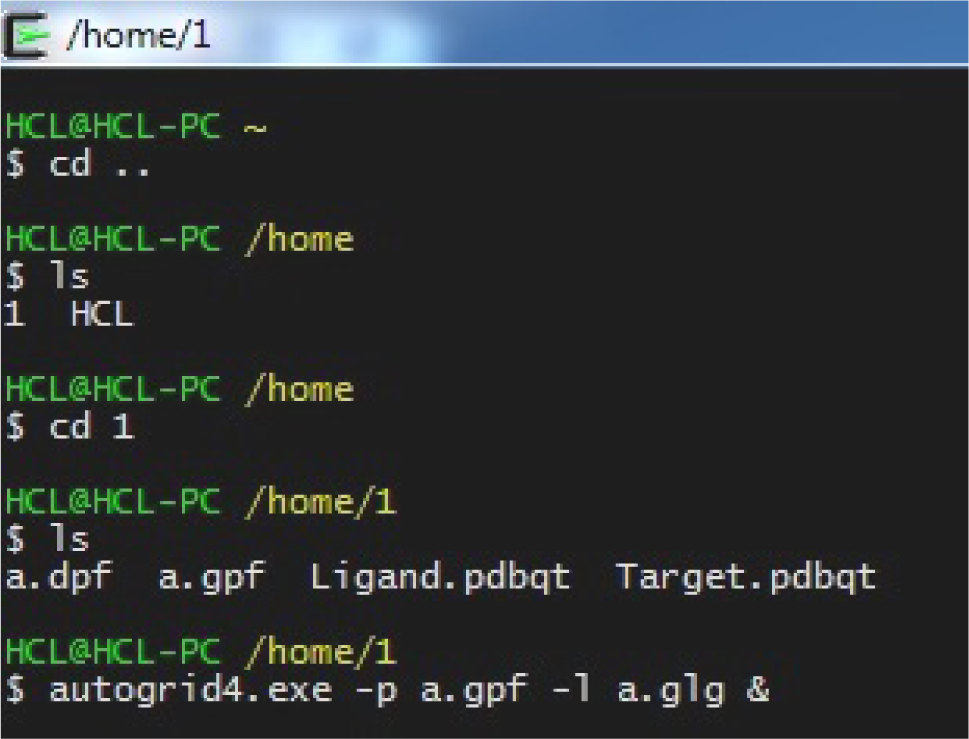
(cd..)cd<space>..
(ls)ls<space>
(cd 1) cd<space>1(or foldername)<space>
(ls)ls<space>
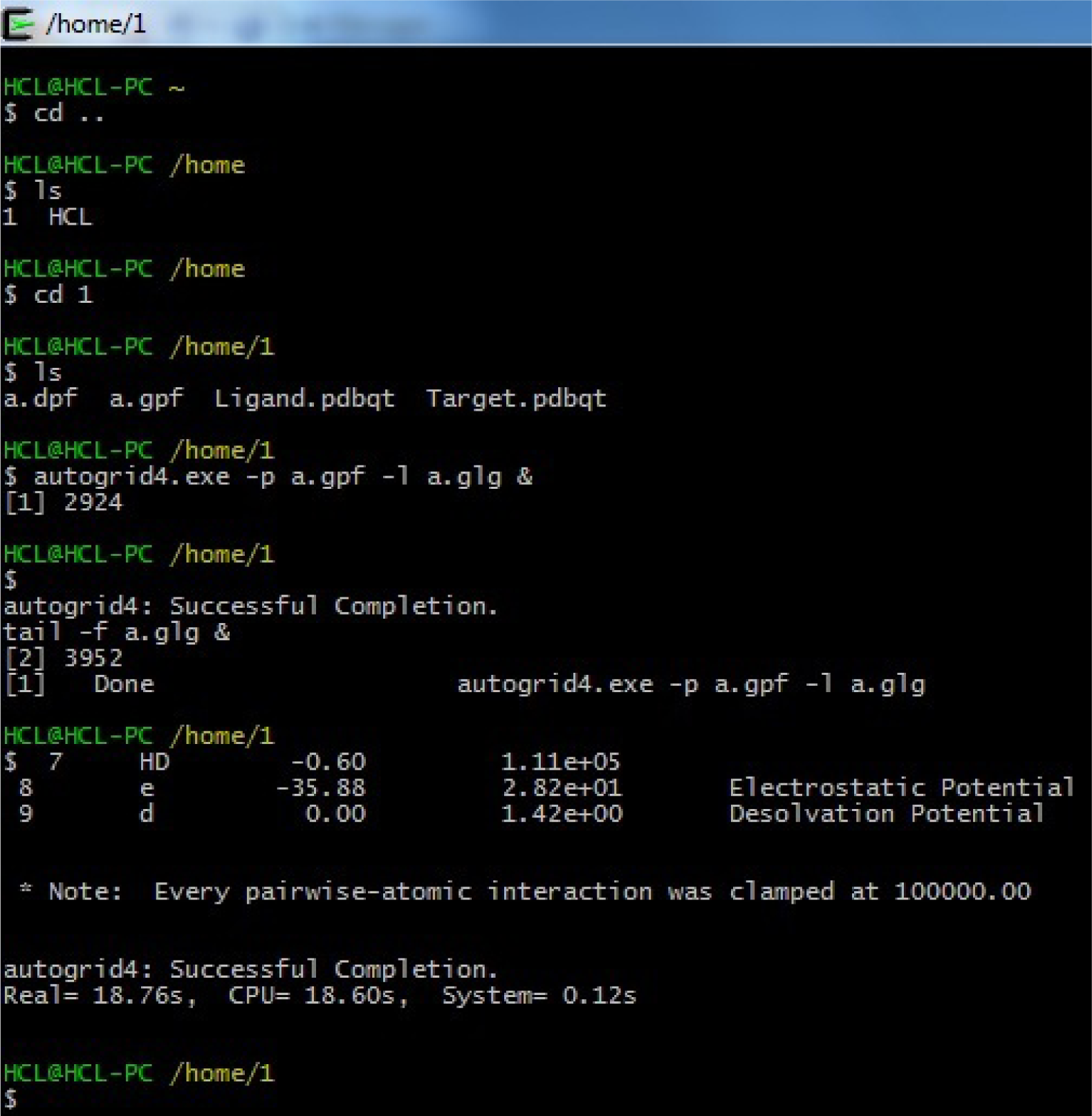
(autogrid4.exe -p a.gpf -l a.glg &)
autogrid(tab)<space>-p<space>a.gpf<space>-l<space>a.glg&
(Fig. 34)
(tail -f a.glg &) tail<space>-f<space>a.glg<space>&
(Fig. 35)
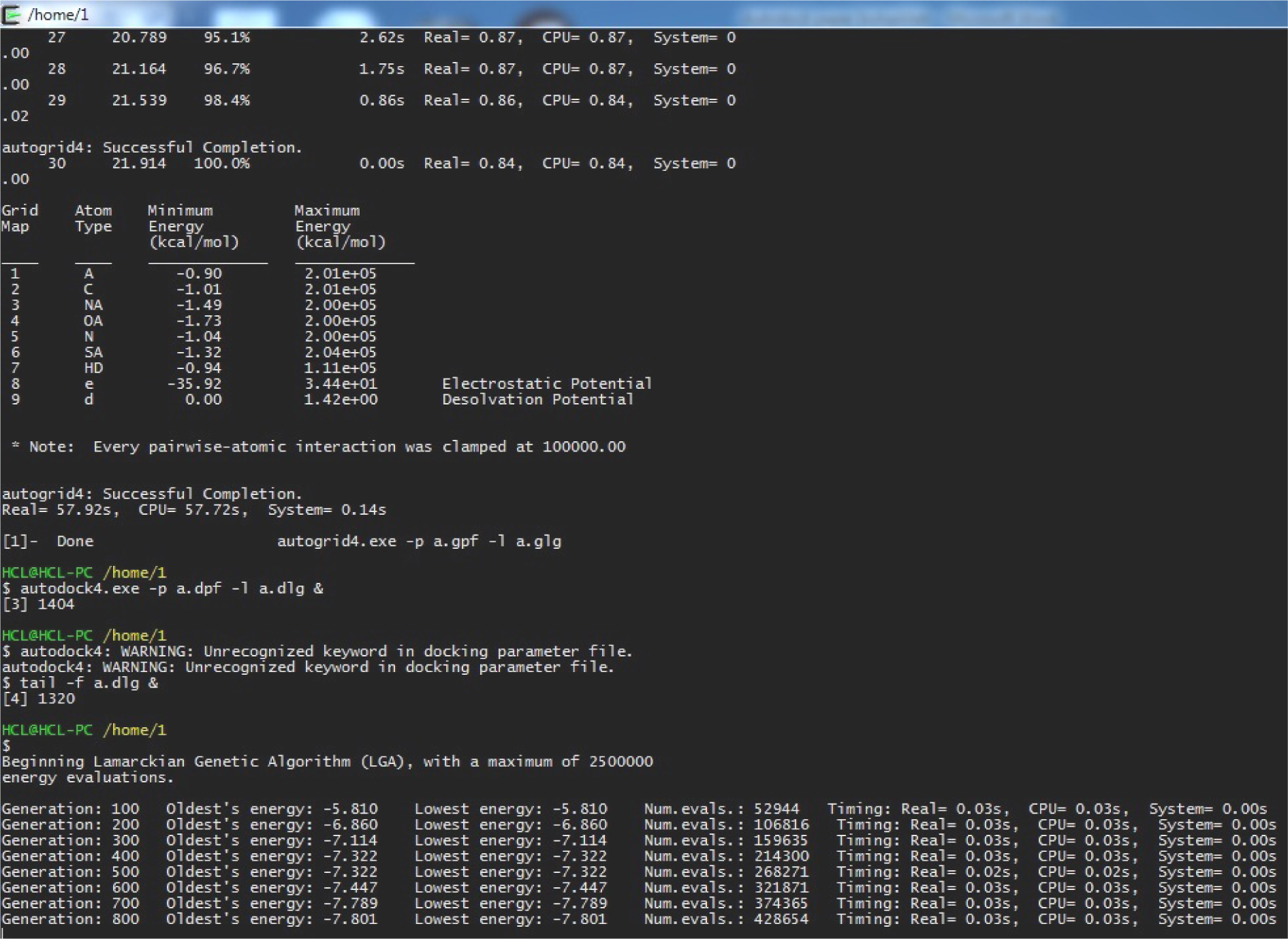
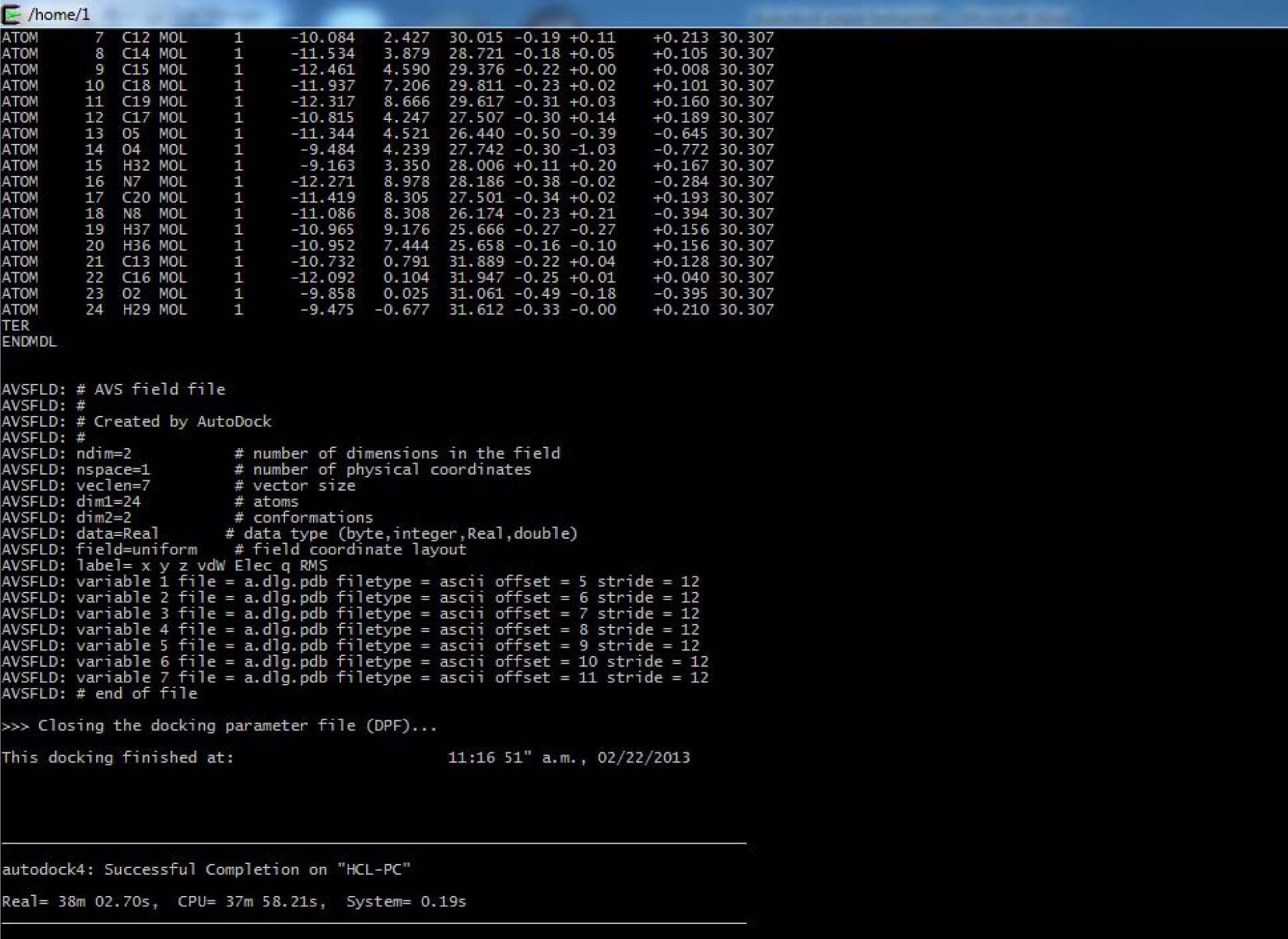
(autodock4.exe -p a.dpf -l a.dlg &)
autodock(tab)<space>-p<space>a.dpf<space>-l<space>a.dlg&
(tail -f a.dlg &) tail<space>-f<space>a.dlg<space>&
(Fig. 36)
(After Successful Completion)
(Fig. 37)
Copy Target.pdb file in C:\Cygwin\ home\1
(Fig. 38)
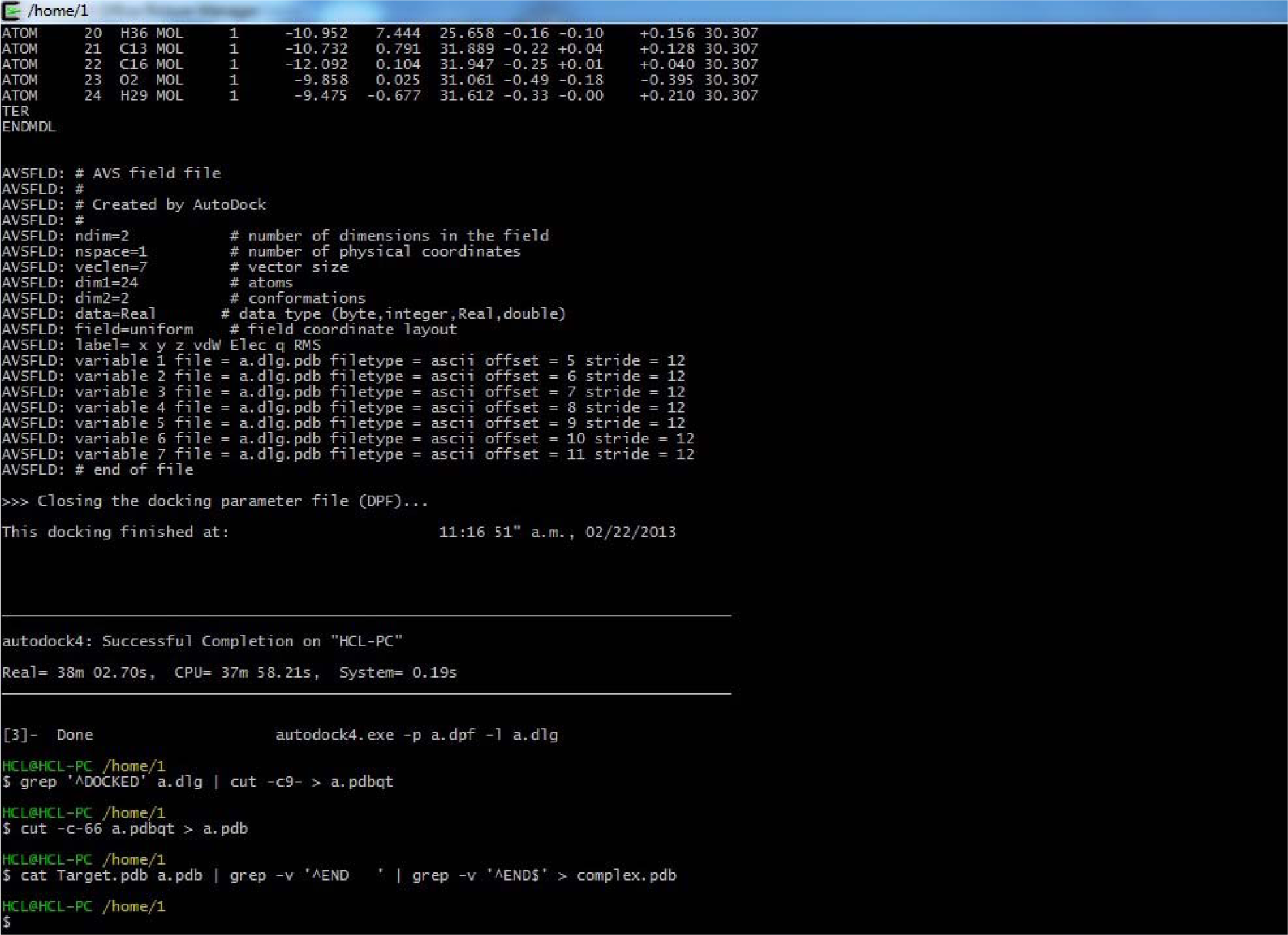
Copy and Paste the following commands in Cygwin Window and press enter after each command:
(grep '^DOCKED' a.dlg | cut -c9- >a.pdbqt)
(cut -c-66 a.pdbqt> a.pdb)
(catTarget.pdb a.pdb | grep -v '^END ' | grep -v '^END$' > complex.pdb)
(Fig. 39)
- Close Cygwin Window
- Click OK
4 Analyzing results and Retrieving Ligand-Enzyme interaction complex .pdb
4.1 Analyzing Results
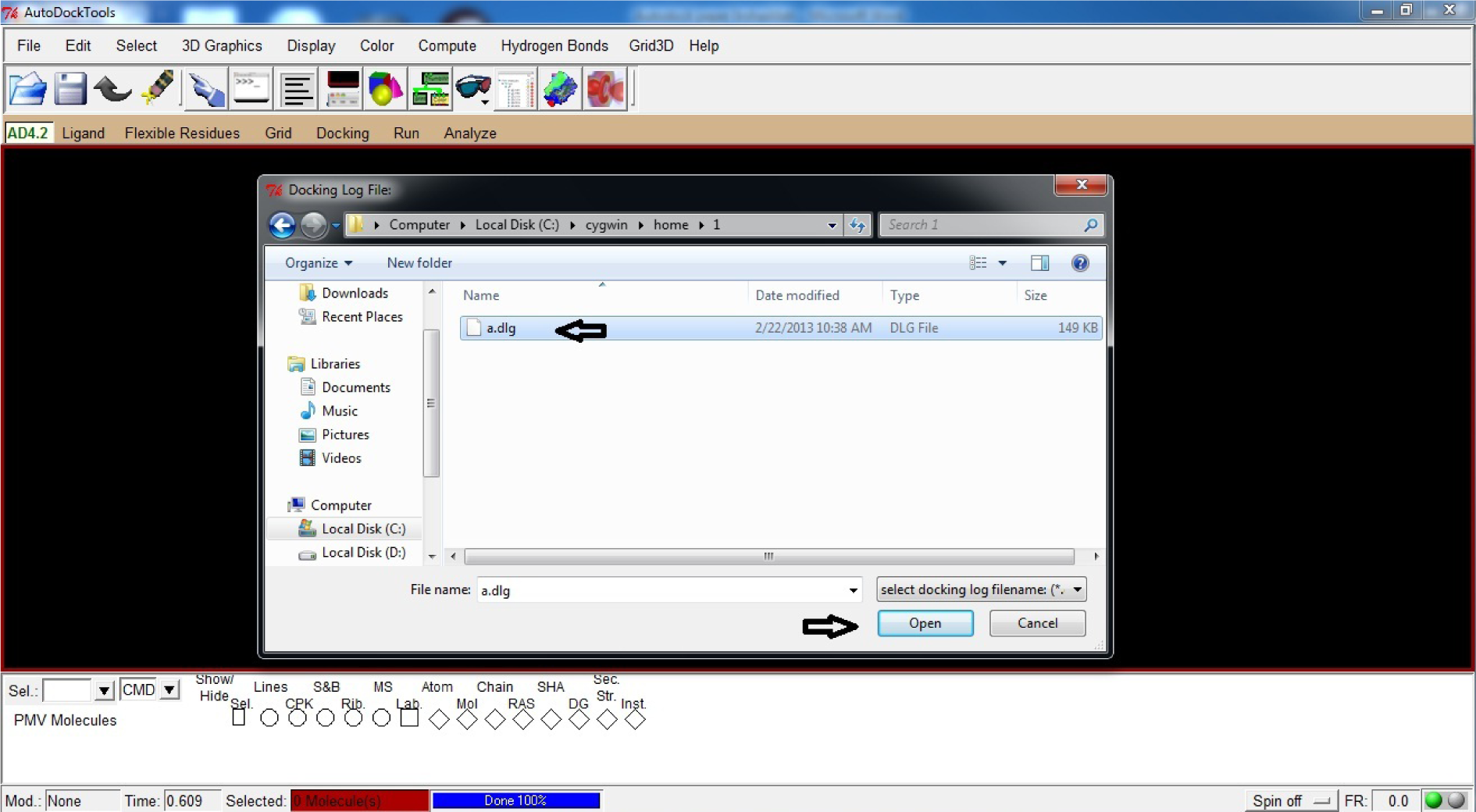
- Open AutoDock
- Click Analyze
- Click Docking
- Click Open
- Select a.dlg
- Click Open
(Fig. 40)
- Click OK
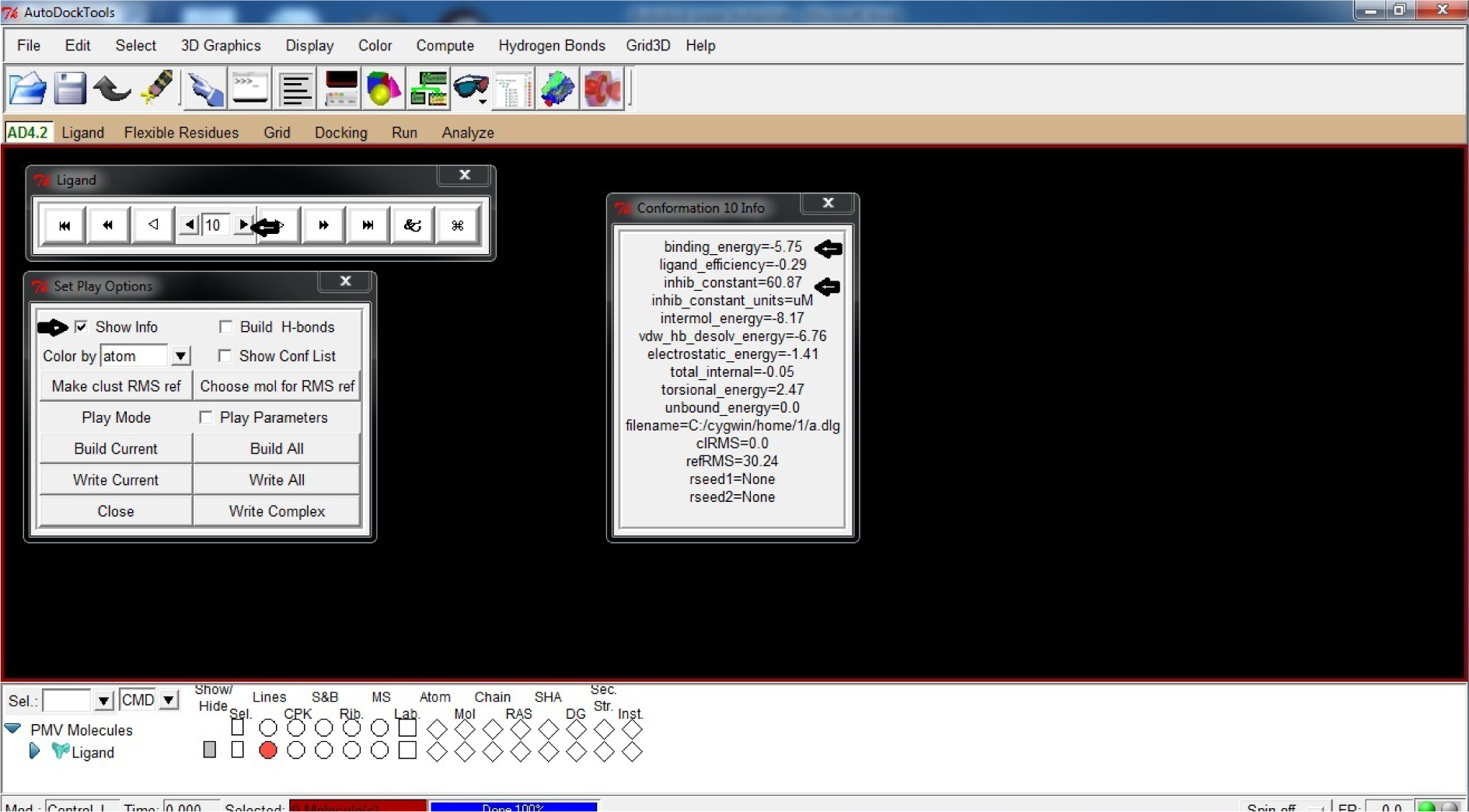
- Again Analyze
- Click Conformations
- Click Play
- Click &
- Click show information
- Click this sign ► to observe each conformation from 1 to 10
Note the confirmation showing best down binding energy and inhibition constant
(*In our case 10 conformation was best with binding energy (ΔG) as -5.75 and inhibition constant (Ki) as 60.87 µM)
(Fig. 41)
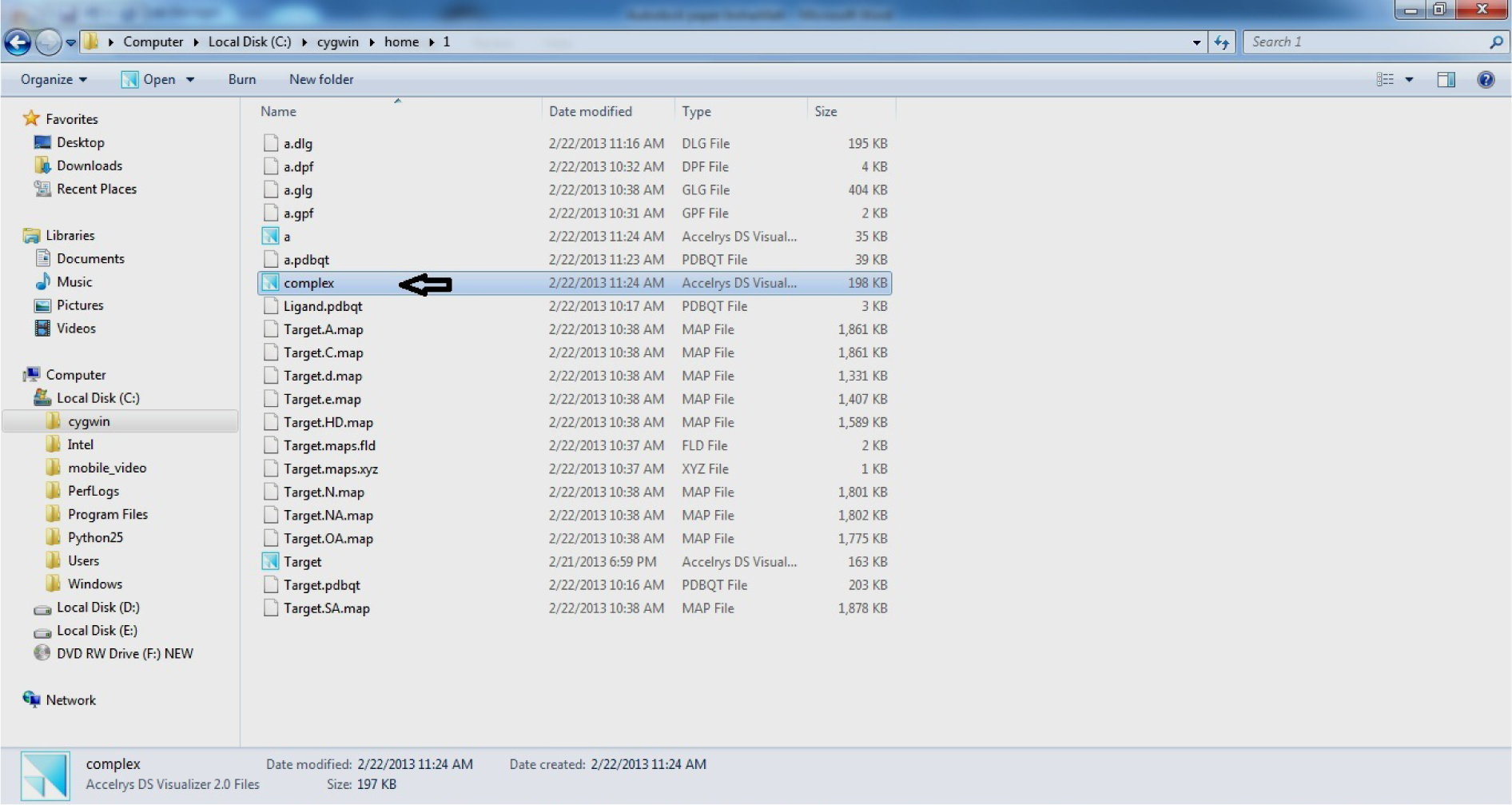
4.2 Retrieving Ligand-Enzyme interaction complex .pdb
- Open C drive
- Open Cygwin
- Open home
- Open 1
- Open complex.pdb in Discovery Studio Visualizer
(Fig. 42)
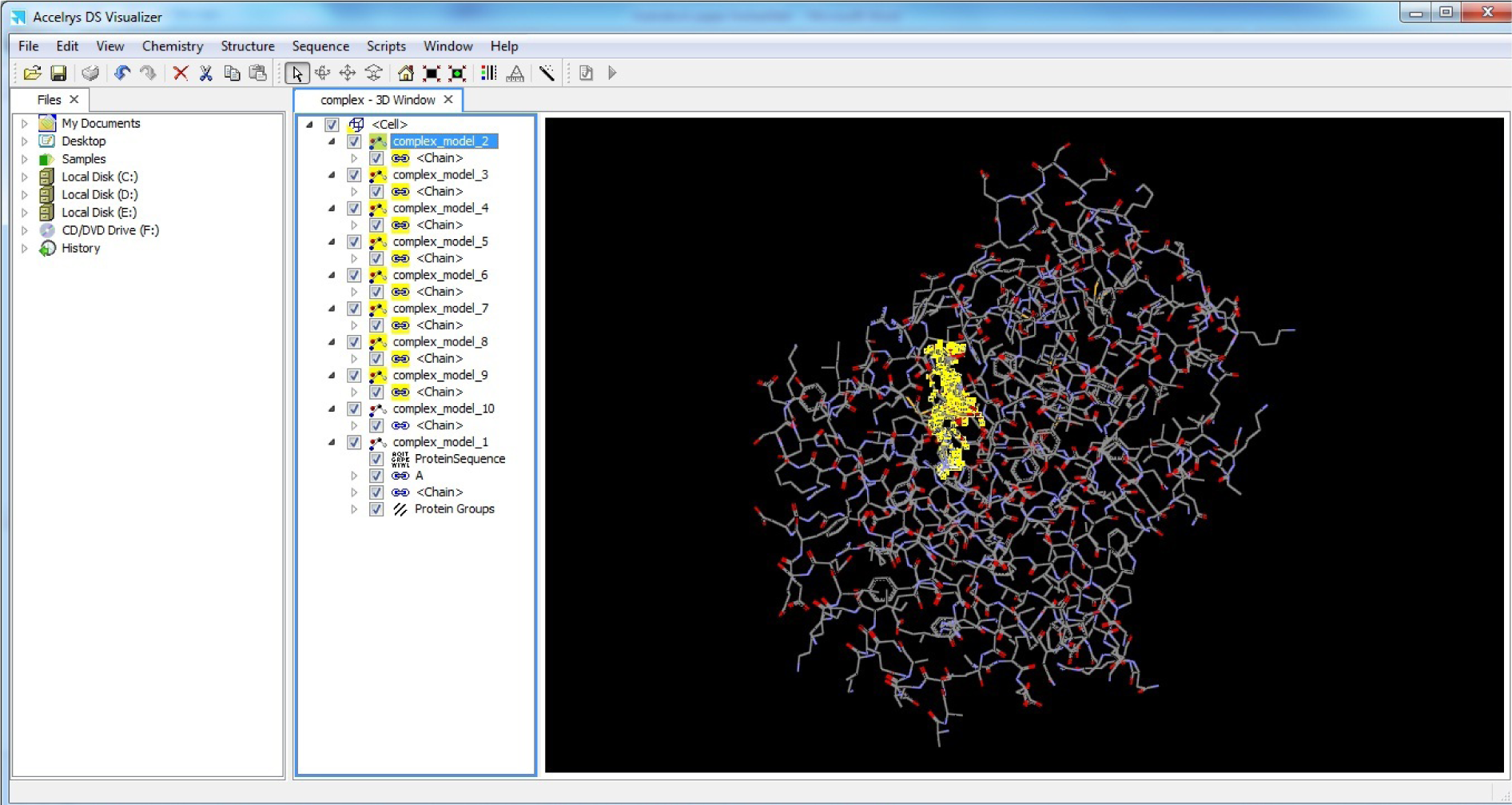
Select all other complexes and delete them except the best
(*In our case Complex model 10 was best as conformation 10 was showing best results in our case).
(Fig. 43)
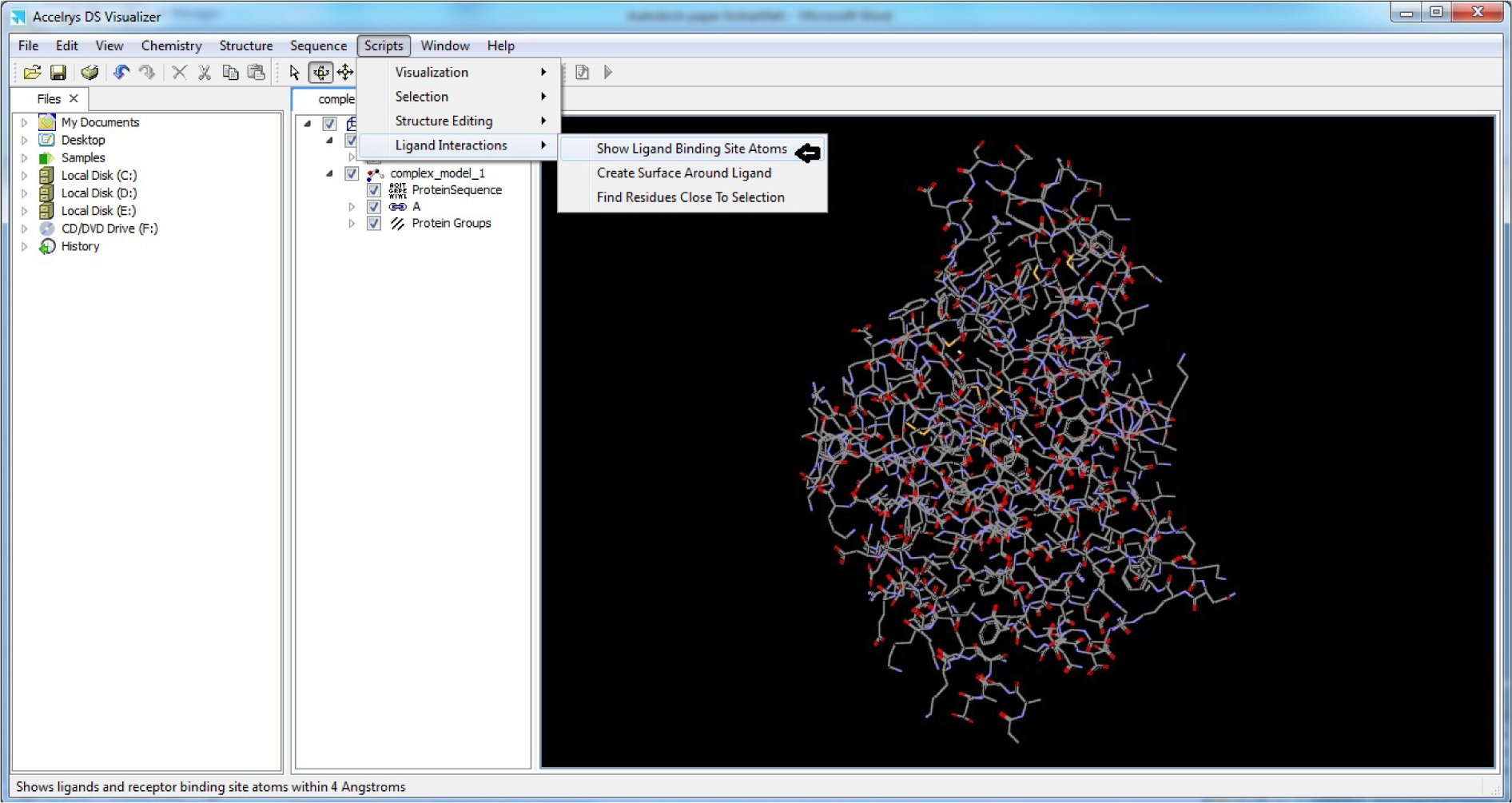
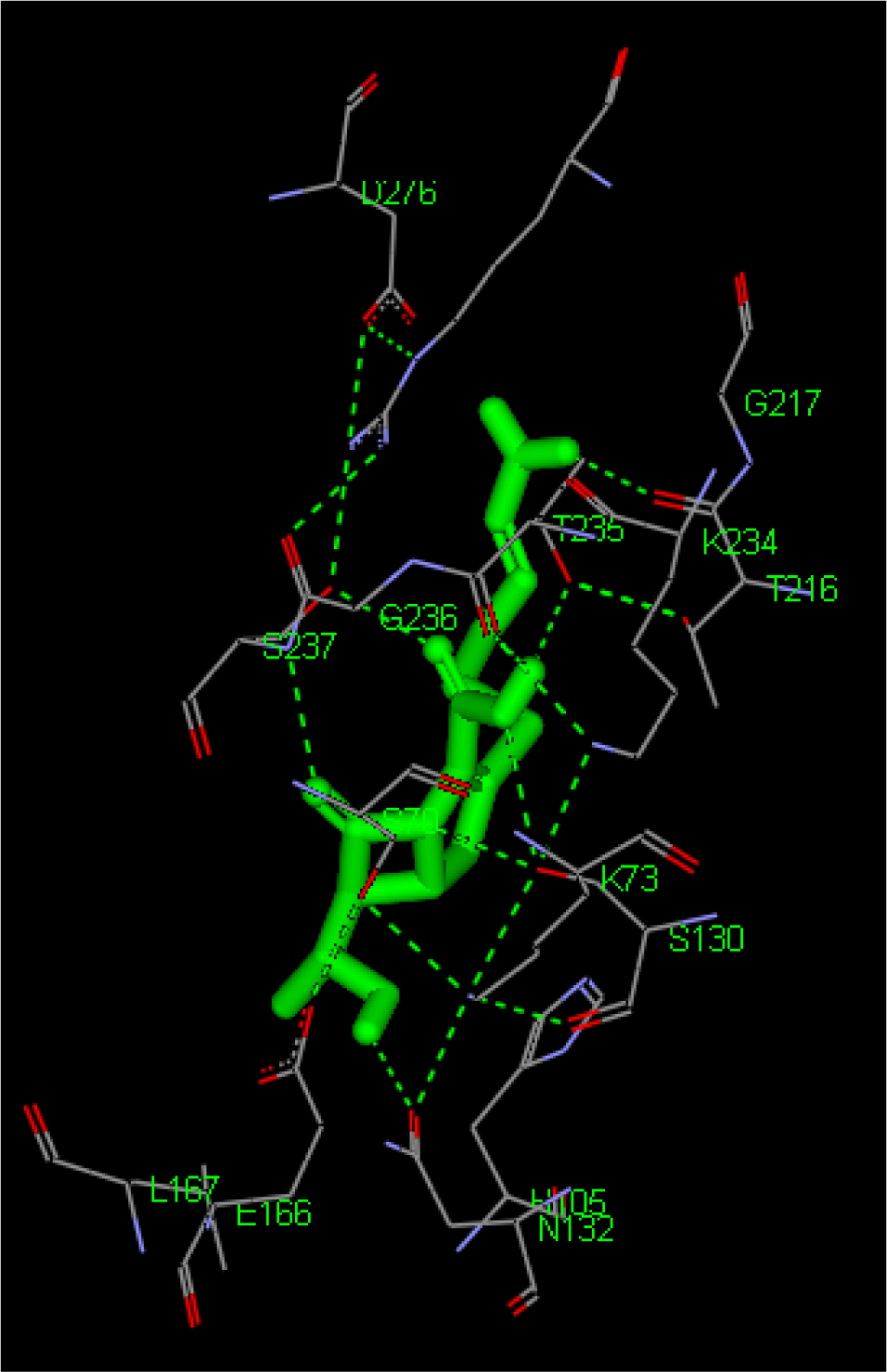
- Click Scripts
- Click Ligand Interactions
- Click Show Ligand Binding Site Atoms
(Fig. 44)
- Right Click on Complex
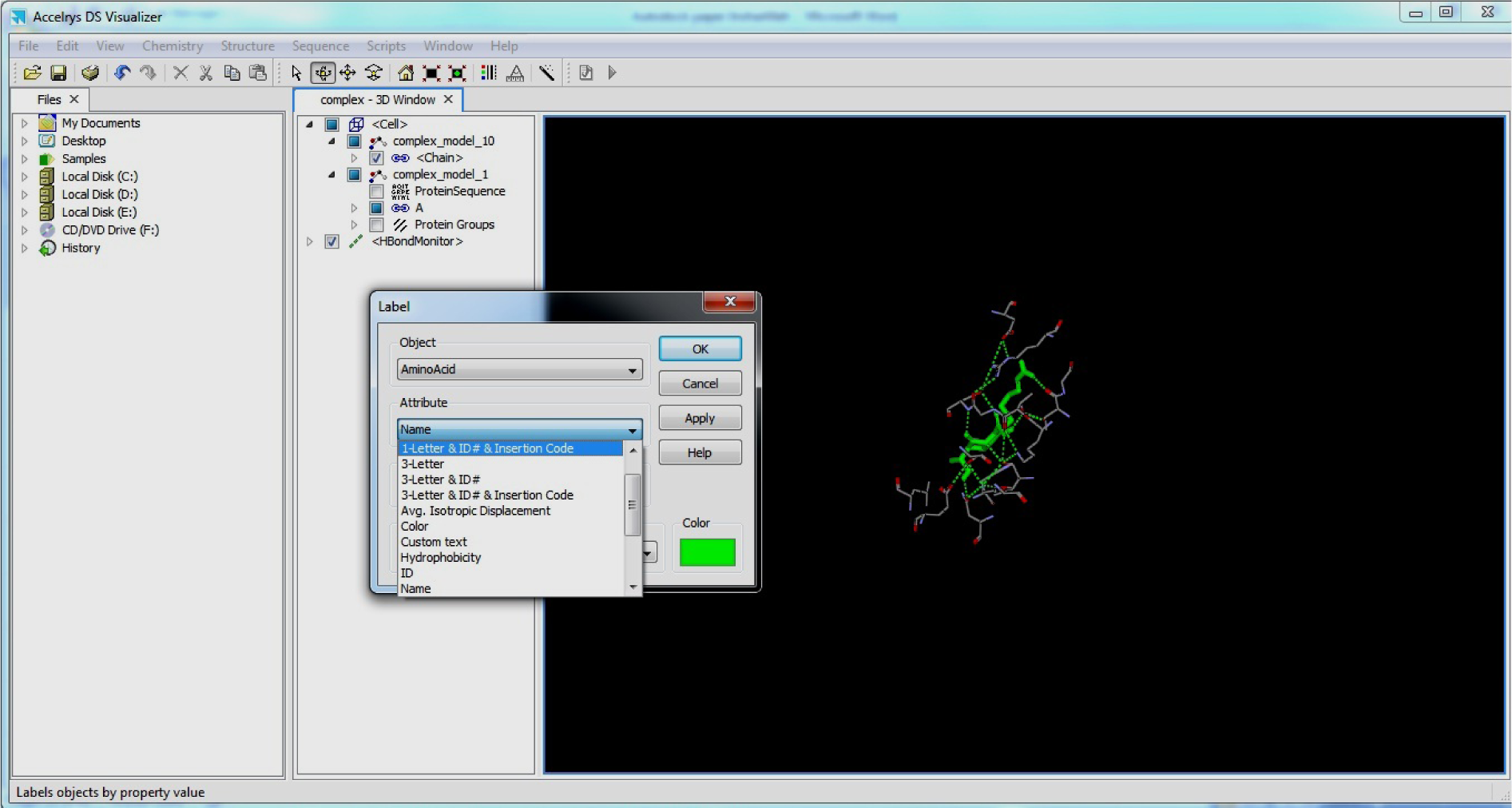
- Click Label
- Select Object: AminoAcid
- Select Attributes: 1 Letter & ID insertion code
- Click OK
(Fig. 45)
- Save as Image files
(Fig. 46)
Conclusion
AutoDock is a popular non-commercial docking program that docks a ligand to its target protein and performs well (accurate and computationally fast). In this paper we propose an easier user-friendly docking protocol for docking ligands with target protein that utilizes AutoDock and Cygwin for docking operations. Our protocol provides a detailed outline and advice for use of AutoDock, AutoDock Tools, its graphical interface and to analyze interaction complexes using computational docking. The example of a docking experiment between Imipenem-hydrolyzing beta-lactamase SME-1 (an enzyme) and Imipenem (a ligand) using AutoDock 4.2/ADT has been given. Our sincere aim is to spread knowledge and make scientific research accessible to researchers who could not afford to buy software or pay high subscription fees of online docking servers. With due confidence, this is our humble claim that a researcher with no previous background in bioinformatics research would be able to perform molecular docking using AutoDock 4.2 program by following stepwise guidelines given in this article.
Acknowledgements
The authors are thankful to all the scientists of this world who possess a burning desire to share their knowledge and skills with the entire world free of charge and solely for the benefit of mankind and expect its reward from Allah alone. We extend sincere thanks to the inventors of ‘AutoDock’.
References
[*] Corresponding Author:
Dr., Assistant Professor, Shazi Shakil, Department of Bio-engineering, Integral University, Lucknow, UP, India-226026; Phone: 0522-2890812, 2890730, 3296117, 6451039; Fax: 0522-2890809; Mobile: +91-8004702899, eMail: shazibiotech@gmail.com