Research article
DNA and chromosomal damage in coronary artery disease patients
Mohd Akbar Bhat1[*], Naresh Mahajan1, Gursatej Gandhi1
1Department of Human Genetics, Guru Nanak Dev University Amritsar 143005EXCLI J 2013;12:Doc872
Abstract
DNA and chromosomal damage in peripheral blood leukocytes of patients with coronary artery disease (CAD) were investigated by using the single cell gel electrophoresis assay /comet and cytokinesis- block micronucleus (CBMN) assays, respectively. The case-control study comprised patients with CAD (n = 46; average age 53.0 ± 1.27 y) undergoing treatment at local hospitals, and healthy age-and sex-matched controls (n = 19; average age 54.21 ± 0.91 y) from the general population. The results of the comet assay revealed that the mean values of DNA damage were significantly (p < 0.001) higher in CAD patients than in controls (Tail DNA% 11.55 ± 0.38 vs. 5.31 ± 0.44; Tail moment 6.17 ± 0.31 vs. 2.93 ± 0.21 AU; Olive tail moment 3.52 ± 0.23 vs. 1.25 ± 0.11 AU). The mean values of chromosomal damage were also significantly higher (p < 0.001) in CAD patients than in controls (Binucleated cells with MN- 28.15 ± 1.18 vs. 18.16 ± 2.59; micronuclei 29.52 ± 1.21 vs. 18.68 ± 2.64, respectively) while nuclear division index (1.48 ± 0.01 vs. 1.63 ± 0.01) was significantly higher (p < 0.001) in controls. The results of the present study indicate that coronary artery disease patients had increased levels of both, unrepaired (DNA) and repaired (chromosomal) genetic damage which may be a pathological consequence of the disease and/or the drug-treatment. This accumulation of DNA/chromosomal damage is of concern as it can lead to the development of cancer with increased chances of morbidity and mortality in the CAD patients.
Keywords: coronary artery disease, DNA damage, chromosomal damage, comet assay, CBMN assay
Introduction
Coronary artery disease (CAD) is one of the most common causes of death and disability worldwide (Zaman et al., 2000[50]). Genetic predisposition and interaction with a host of environmental (endogenous and exogenous) factors can cause the disease. Among the risk factors predisposing to CAD are hypertension, obesity, hyperlipidemia, stress, smoking, age, gender and family history (Kasap et al., 2007[24]). Oxidative stress has also emerged as a major risk factor (Yung et al., 2006[49]) in CAD arising through an imbalance between oxidants and antioxidants (Nojiri et al., 2001[36]). The oxidative modification of low density lipoproteins (LDL) also plays a causative role in the pathogenesis of coronary artery disease (Chisolm and Steinberg, 2000[11]). Furthermore, consequences of oxidative stress can cause damage to genomic integrity as atherosclerotic plaques have been reported to exhibit extensive genetic damage (Martinet et al., 2002[31]). In fact, reports have also documented the contribution of DNA damage to the development and progression of coronary artery disease (Andreassi, 2003[1]).
There has been an increase in heart disease in Punjabi population which may be attributed to economic surplus and change in dietary and life-style patterns (Anonymous, 2013[4]). The changeover from extensive agrarian activities to a sedentary / near sedentary life-style and the adaptation of the western dietary habits across the lower, upper middle and upper socioeconomic status classes, have resulted in increased general and abdominal obesity (WHO, 2003[48]). As no studies have come to attention in Punjabi populations associating CAD with genomic damage, in the present study, adults (CAD patients and healthy controls) have been assessed for chromosomal and DNA damage in their peripheral blood leukocytes (PBL). Correlation of genomic damage has further been studied with various risk factors viz. lipid profile, hypertension, diabetes, obesity parameters (body mass index, waist-hip ratio, waist circumference) and oxidative stress (lipid peroxidation, total oxidant status, total antioxidant capacity, oxidative stress index).
For genetic damage assessment, chromosomal damage in peripheral blood lymphocytes using the Cytokinesis-Block micronucleus (CBMN) assay and DNA damage in PBL using the single cell gel electrophoresis (SCGE/comet) assay have been carried out. The CBMN assay is a well known cytogenetic test-method to assess the genetic effects of spontaneously and mutagenically- induced DNA damage (Fenech, 2007[17]) and the comet assay is a sensitive biomarker of oxidative stress and DNA damage (Balasubramanyam et al., 2010[6]).
Materials and Methods
Subjects
The study proposal was approved by the Institutional Ethics Committee and voluntary written informed consent was taken from all study participants (n = 65). Patients with diagnosed CAD were contacted from local hospitals through the cardiologists. Exclusion criteria included CAD patients with renal, liver or thyroid disorders, those with negative results on the treadmill test, and those not belonging to the Punjabi population. The control group included healthy adults with no past or present history of heart or any disease and no exposure history, matched for gender, population group and socioeconomic status.
Data and sample collection
The demographic and disease-related information for each patient was documented on a pre-designed questionnaire. Anthropometric measurements (height, weight, hip circumference, waist circumference) were taken following standard methodology (Weiner and Lourie, 1981[46]). Derived obesity measures were Body Mass Index (BMI) and Waist Hip Ratio (WHR). Using the criteria of WHO (2004[47]) for BMI, the participants were classified as obese or non-obese regarding general obesity, and cut-offs as per Snehalatha et al. (2003[42]) for WHR and waist circumference (WC) values for abdominal obesity. Physiometric measurements (using a mercury sphygmomanometer and stethoscope) were taken after the subject had rested for ten minutes as per the recommendations of American Heart Association (Kirkendall et al., 1981[26]). The average of three measurements were taken as the blood pressure values. The mean arterial blood pressure (MBP) was calculated from systolic and diastolic measurements (Pérusse et al., 1989[38]). Venous blood (5 ml) was then drawn from each participant and processed for the CBMN and SCGE assays.
The Cytokinesis-Block Micronucleus (CBMN) assay: - Short term lymphocyte cultures were set up (Moorhead et al., 1960[34]) and the CBMN protocol of Fenech (2000[18]) was followed. Briefly, the leukocyte-rich plasma (2 ml) was added to 8 ml of RPMI 1640 medium (HiMedia, India). The lymphocytes were stimulated to divide by addition of Phytohaemagglutinin-M (0.5 µg/ml PHA-M, Biological Industries, Israel). The cultures were incubated for 72 h at 37 °C; after 44 h cytokinesis was blocked by the addition of cytochalasin-B (6 µg/ml, Sigma) and on completion of incubation of 72 h at 37 °C, the cultures were harvested. Following centrifugation (800 g, 10 min), the pellet was fixed in chilled fixative (3 methanol: 1 glacial acetic acid, Qualigens, India and SRL, India, respectively) for 30 min. After centrifugation and two-three washings of the pellet, the cell suspension was used to prepare the slides. The air-dried slide preparations were stained with 5 % Giemsa (HiMedia, India) for 10-20 minutes. For each sample, two slides were prepared and coded. A total of 2000 binucleated cells (1000/slide) were scored for the presence of micronuclei (MN) and micronucleated cells following the criteria of Fenech (2007[16]). Random observations by another scorer were done to verify the observations. The Nuclear Division Index (NDI), which is an index of cytostatic effects, was calculated (Eastmond and Tucker, 1989[14]) by using the formula:
NDI = (M1+2M2+3M3+4M4)/N
where M1-M4 represent the number of cells with 1-4 nuclei and N is the total number of cells scored.
The SCGE/Comet assay: The Trypan-Blue Dye Exclusion test for cell viability was performed (Strober, 2001[45]) and samples with > 95 % cell viability were processed for the comet assay.
The leukocytic DNA damage was ascertained using the alkaline comet assay (Singh et al., 1998[41]). In brief, on pre-coated slides with 1 % normal melting point agarose (NMPA; SRL, India), a second layer containing 30 µl of blood sample mixed with 110 µl of 5 % low melting point agarose (LMPA, SRL, India) was poured followed by the sandwiching with a third layer of 120 µl of 5 % LMPA after the second layer had set. Cells were lysed in freshly prepared cold lysing solution (2.5 M NaCl, 100 mM Na2 EDTA, 10 mM Tris Hcl, pH 10.0; 1 % Triton X-100 with 10 % DMSO added just before use) for at least one hour to allow DNA unfolding. Then the slides were placed in a horizontal electrophoresis chamber, covered with freshly prepared alkaline electrophoresis buffer (0.3 M NaOH, 1 mM NO2 EDTA, pH > 13) at 4 °C for unwinding and expression of alkali-labile sites (30 min) and subsequently electrophoresed (25 V, 300 mA, 0.7-1.0 -v/cm, 25 min). Slide preparations were then washed with neutralizing buffer (0.4 M Tris HCl pH 7.5) to remove alkali and detergents, and left for drying. The slides were silver stained (Nadin et al., 2001[35]) and coded. Scoring was done using the freely available CASP (Comet Assay Software Programme, 3.1.2 version). For this, images were captured with an Olympus camera (E420, Tokyo, Japan) fitted on an Olympus microscope (CX41, Tokyo, Japan). A total of 100 cells per sample were scored for per cent tail DNA, Tail Moment and Olive Tail Moment as indices of DNA damage.
Statistical analysis
Statistical analysis was performed using the Statistical Package for the Social Sciences (SPSS, version 16.0 for Windows 7). The results on various observations are expressed as mean ± S.E.M. The data showed normal distribution and therefore parametric tests were performed. Differences between controls and patients for genetic damage indices were analyzed by the Student's t-test. Demographic variables of patients and control groups were analyzed using the Chi-square test. The association between DNA/chromosomal damage and other variables was assessed by the Pearson's correlation test. The independence of these associations was tested by multiple linear regression analysis. For all calculations, the level of significance was taken as p < 0.05.
Results
The demographic and clinical characteristics of patients and controls are presented in Table 1(Tab. 1). CAD patients had been diagnosed by the cardiologists on the basis of clinical symptoms, ECG findings, treadmill test and echocardiography. There were 26 males (mean age 49.62 ± 1.73 y) and 20 females (mean age 57.30 ± 1.31 y) in the patient group. The control group included 13 males (mean age 53.54 ± 1.17 y) and 6 females (mean age 55.67 ± 1.33 y). On the basis of BMI, there were 14 obese patients (BMI ≥ 25.0 kg/m2) while the WHR revealed that 26 male and 20 female patients were obese as per the cut-off values given by Snehalatha et al. (2003[42]). On the basis of the waist circumference cut-offs (Snehalatha et al., 2003[42]), 23 male (> 85 cm) and 19 female (> 80 cm) patients had abdominal obesity. Alcohol drinking was observed in 52.20 % male patients and in 31.60 % male healthy controls. There were 28.30 % patients with diabetes mellitus and 56.50 % with hypertension. Non-vegetarian diet was preferred by 58.70 % of the patients and 52.60 % of the controls and sedentary life style was observed by 71.70 % CAD patients and only by 36.80 % of controls. Regarding medication in the patients group, 67.40 % were taking Aspirin (anti-inflammatory), 58.70 % Lasix (diuretic), 52.20 % Metoprolol (beta-blocker) and 47.80 % were taking Ramipril (ACE-inhibitor). Socioeconomic status (SES) was inferred according to the criteria given by Kumar et al. (2007[27]). There were almost equal percentage of patients (15.20 %) and controls (15.80 %) belonging to upper SES. Similarly, 76.10 % patients and 13.70 % controls belonged to upper-middle SES, while 8.70 % among patients and 10.50 % among controls had lower-middle SES. Information about the history of disease revealed that date-of-onset/diagnosis ranged from 35-72 y (average 53.09 ± 1.3 y) and the patients were on medication from 1-15 y (average 5.04 ± 0.6 y).
On Chi-square analysis it was observed that the demographic variables of patients and controls matched for age, gender, alcohol drinking, diet preference, socioeconomic status, WHR (men) and waist circumference (women), while BMI, diabetes, dyslipidemia, physical activity, WHR (women), waist circumference (Men), blood pressure, MBP and medication showed differences being characteristic of patients. In Table 2(Tab. 2), the information on genetic damage indices in patients and controls is presented. The Trypan-Blue Dye Exclusion test had revealed 95.60 - 99.80 % cell viability of all samples. The comet parameters used as metrics of DNA damage were Tail moment (TM = Tail DNA% x tail length in arbitrary units, AU), Tail DNA% (TD = amount of DNA in comet tail in percentage) and Olive Tail Moment (OTM = tail mean-head mean x % DNA in tail in AU). A comparison between patients and controls for these DNA damage indices revealed that the values were significantly (p < 0.001) higher in patients compared to those in controls. However no gender differences for these values were observed among patients and hence the respective data were pooled for genders in both, the control and patient groups. Separate analysis also showed highly significant differences (p < 0.001) for both, male and female patients from their control counterparts for DNA damage indices.
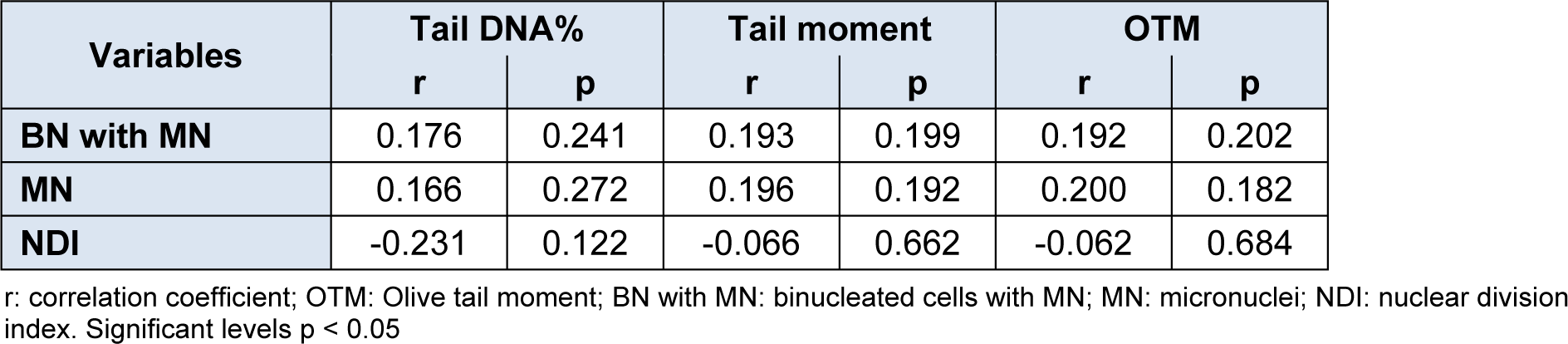
For the assessment of chromosomal damage, the binucleated cells with micronuclei (BNMN), number of micronuclei in binucleated cells (MN) and the nuclear division index (NDI) were studied. Statistical analysis revealed that the BNMN and MN values were significantly (p < 0.001) higher and NDI values lower in patients than in controls. As observed for DNA damage, no gender differences for chromosomal damage were observed among patients and controls, though values in male and female patients for BNMN and MN were significantly higher (p < 0.001) in patients while NDI values were significantly (p < 0.001) higher in controls. On correlation and association analyses (Table 3(Tab. 3)), significant positive correlation (correlation coefficient = r) was observed between BNMN and diabetes mellitus (r = 0.297, p = 0.045) and between NDI and MDA levels (r = 0.296, p = 0.045) and with age (r = 0.571, p = 0.000). Positive correlation was observed between tail DNA % and diabetes mellitus (r = 0.442, p = 0.004) and with WHR (r = 0.349, p = 0.017) and between tail moment and age (r = 0.312, p = 0.035). On multiple linear regression analysis (Table 4(Tab. 4)), regression coefficient (B) values for age (B = 0.076, p = 0.035), WHR (B = 25.859, p = 0.018) and diabetes mellitus (B = 2.251, p = 0.004) were observed to be independent predictors for DNA damage and NDI for chromosomal damage (B = 0.007, p = 0.001). In order to discern if there was any association between DNA damage and chromosomal damage parameters, correlation analysis was carried out but it revealed no significant associations (Table 5(Tab. 5)).
As oxidative stress and lipid profile biomarkers of this study group had earlier been reported to be significantly higher among the patients (Bhat et al., 2012[7]), it was also thought appropriate to ascertain whether there was any correlation (Table 3(Tab. 3)) and/or association (Table 4(Tab. 4)) of these variables with genetic damage. Correlation and regression analyses were hence performed for genetic damage (DNA/chromosomal damage) with oxidative stress parameters of malondialdehyde (MDA), total oxidant status (TOS), total antioxidant status (TAS) and oxidative stress index (OSI) and also with lipid profile biomarkers (total cholesterol, High density lipoprotein cholesterol (HDL-C), Triglycerides, very low density lipoprotein (VLDL) and low density lipoprotein (LDL). These showed neither correlation nor association with genetic damage except HDL-C and MDA levels which showed positive correlation (r = 0.322, p = 0.029; r = 0.296, p = 0.046 respectively) and regression (B = 0.259, p = 0.029; B = 0.013, p = 0.046 respectively) with per cent tail DNA and NDI respectively.
Discussion
It has often been reported that genetic damage may be caused by both, extrinsic and intrinsic environments (Gray and Bennett, 2011[19]). The DNA damage can manifest as DNA strand-breaks, alkali-labile sites, incomplete repair sites and cross-links while chromosomal damage appears as chromosomal aberrations (gaps, breaks, translocations, rings, insertions) as well as micronuclei (MN). MN originate from chromosome fragments or whole chromosomes that are not included in the main daughter nuclei during nuclear division (Fenech, 2000[18]). In the present study, the SCGE assay was used to measure the level of DNA damage (unrepaired) and the CBMN assay to measure the level of chromosomal damage (repaired) in patients and controls. The results from the comet assay demonstrated that patients diagnosed with CAD had higher values of percentage of tail DNA (2.1x), Tail moment (3 x) and Olive tail moment (3 x), indicative of higher DNA damage in peripheral blood leukocytes of CAD patients than the normal controls. There were no gender differences for these indices on statistical analysis implying that in this study group, genetic damage levels (both DNA and chromosomal) are similar in males and females among patients and controls.
The results of the present study have shown significant increase in unrepaired and repairable genetic damage in peripheral blood leukocytes/lymphocytes of patients with coronary artery disease. Reports in literature have implicated excessive oxidative stress and inadequate defenses in the pathogenesis of cardiovascular disease (Andreassi, 2003[1]) besides genetic damage in atherosclerotic plaques (Martinet et al., 2002[31]). As oxidative stress has been implicated as a major contributor in the progression of atherosclerosis (Norman et al., 2001[37]), it could also be a causative factor for DNA damage in coronary artery disease as DNA damage occurs frequently in cells exposed to reactive oxygen species (Botto et al., 2001[8]). The CAD patients of the present study also had significantly increased (3 folds) oxidative stress as given in an earlier work (Bhat et al., 2012[7]). This oxidative stress hence could be contributing to the observed DNA and chromosomal damage in CAD patients. As dyslipidemia is an important causative factor for coronary artery disease (Arca et al., 2007[5]), the patients of the present study had earlier been assessed for their lipid profile and had higher levels of triglycerides and total cholesterol and low levels of high density lipoprotein-cholesterol despite treatment (Bhat et al., 2012[7]). The HDL-C and MDA levels showed positive correlation with genetic damage on correlation and regression analyses implying that dyslipidemia also induced oxidative stress (higher MDA levels) and could also be contributing to the observed genetic damage in these patients.
In literature, various studies have demonstrated increased genetic damage in patients with heart disease. Increased production of oxidant free radicals was observed in patients with ischemic heart disease (Logacheva et al., 2001[28]) and with evidence of oxidative DNA damage in cells within the atherosclerotic plaque (Mahmoudi et al., 2006[29]) implying that genomic damage may itself be inducing CAD. In their study on Indian CAD patients, Rajesh et al. (2011[39]) reported increased DNA damage (per cent tail DNA) while Botto et al. (2001[8]) had also reported increased DNA damage levels in patients with coronary artery disease. Demirbag et al. (2005[13]) had also documented that DNA strands breaks, oxidized pyrimidines and altered purines were significantly higher in leukocytes of CAD patients.
The Cytokinesis block micronucleus assay was used for chromosomal damage assessment. The key advantage of the MN assay lies in its ability to detect both, clastogenic and aneugenic events, and to identify cells which have divided once in culture (Fenech, 2002[16]). The results of the present study showed that BNMN (1.5 fold) and MN (1.6 fold) values were significant increased in patients while NDI (a biomarker of cytotoxicity) was significantly higher in controls (p < 0.001) than in patients implying that the cell proliferation was reduced while genomic damage was elevated in patients. There is similarity of these results with those of Ramakrishnan et al. (2011[40]) who have also documented increased MN levels among Indian CAD patients. Increase in BNMN among CAD patients in an Italian population had also been earlier reported (Andreassi et al., 2005[2]). The results of the present study are also supported by those of Botto et al. (2001[8]) who had observed approximately 5.6 folds increase in MN levels in CAD patients while Andreassi et al. (2005[2]) had reported 3.1 folds increase in MN frequency in CAD patients.
The observed DNA damage in patients of the present study could also have been contributed by the action of the medications prescribed to these patients namely, Asprin (anti-inflammatory), Lasix/Furosemide (Diuretic), Metoprolol (beta-blocker) and Ramipril (Ace-inhibitors). Studies on genotoxicity of Asprin have shown positive effects in Bacillus subtilis assay (Kawachi et al., 1980[25]) and invitro in the Chinese hamster lung cells (Ishidate, 1983[22]) though negative effects have also been reported in the Ames test (McCann et al., 1975[32]) and in the Chinese hamster ovary cells (Stich et al., 1981[44]). Recent study on Furosemide by Mondal et al. (2012[33]) has reported DNA strand breaks in mice hepatocytes detected by comet assay. Chromosomal aberrations in Chinese hamster ovary and lung fibroblast cells and in mouse germ cells have also been reported (Jameela et al., 1979[23]; Bucher, 1989[10]), yet the genotoxic effects of Furosemide are not well characterized (Bucher, 1989[10]; Brambilla et al., 2006[9]). Recently, on performing mutagenecity testing (Ames test, Comet assay) the effects of degradation products of metoprolol revealed positive genotoxicity (Sojic et al., 2012[43]). Therefore the genetic-damaging effects of these drugs can not be ruled out. Furthermore, the carcinogenicity of cardiovascular drugs, including diuretics, beta blockers, calcium antagonistics and ACE-inhibitors has also been rebated upon (Grossman et al., 1999[20]) and requires further research.
The significantly increased DNA and chromosomal damage observed in the PBL of CAD patients of the present study is of concern as genetic damage is an early indicator of malignancy (Andreassi et al., 2007[3]; Manikantan et al., 2010[30]) and can also promote ageing and neurodegenerative diseases (Farooqui and Farooqui, 2009[15]; Muralidhar et al., 2012). Hence there is a need for preventive management for these aspects in patients with coronary artery disease.
Acknowledgement
The authors are very thankful to Dr. Michael Fenech (CSIRO Human Nutrition, Adelaide, Australia) for providing Cytochalasin-B. The present study was carried out with support out of the departmental UGC-SAP grant.
References
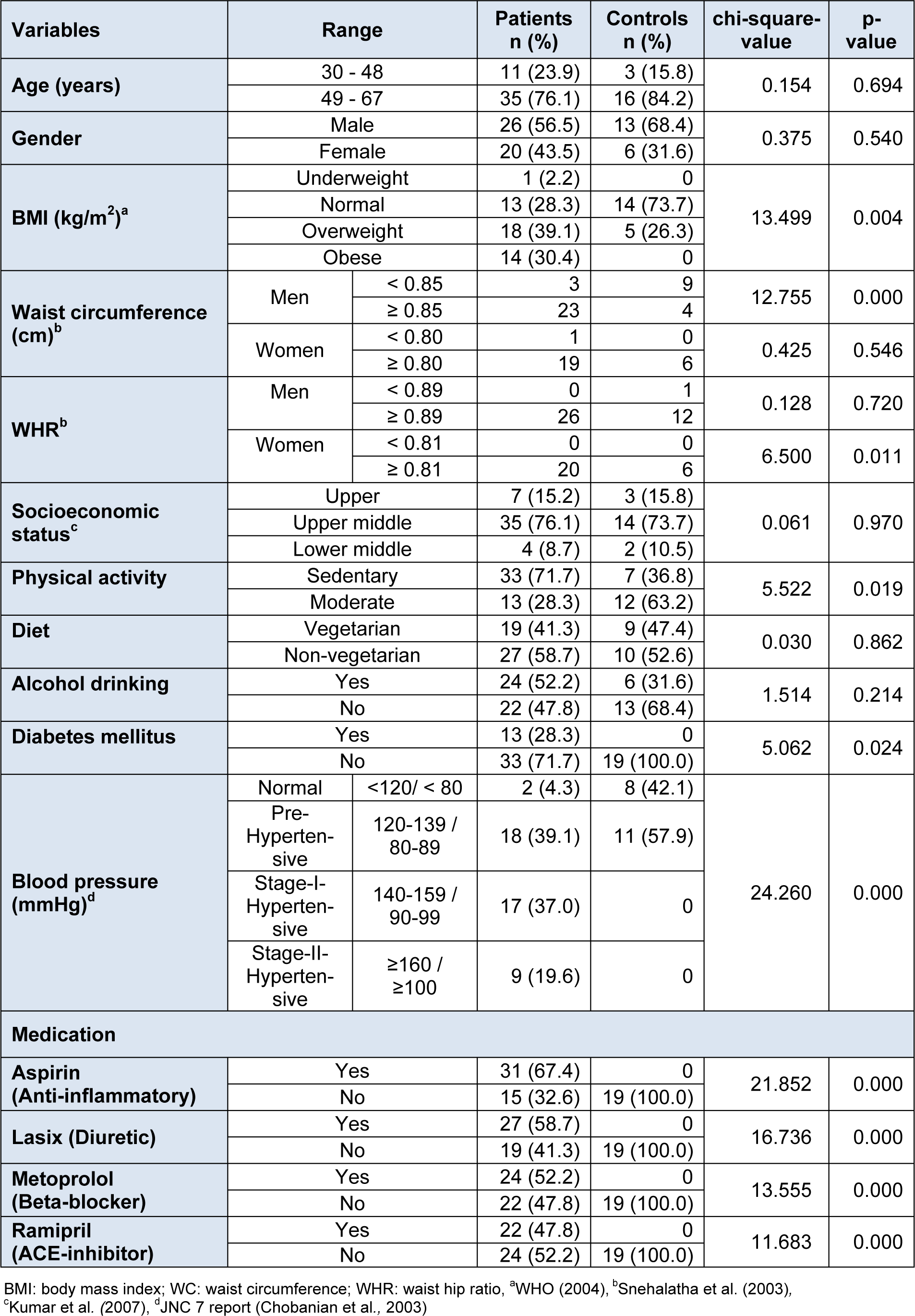
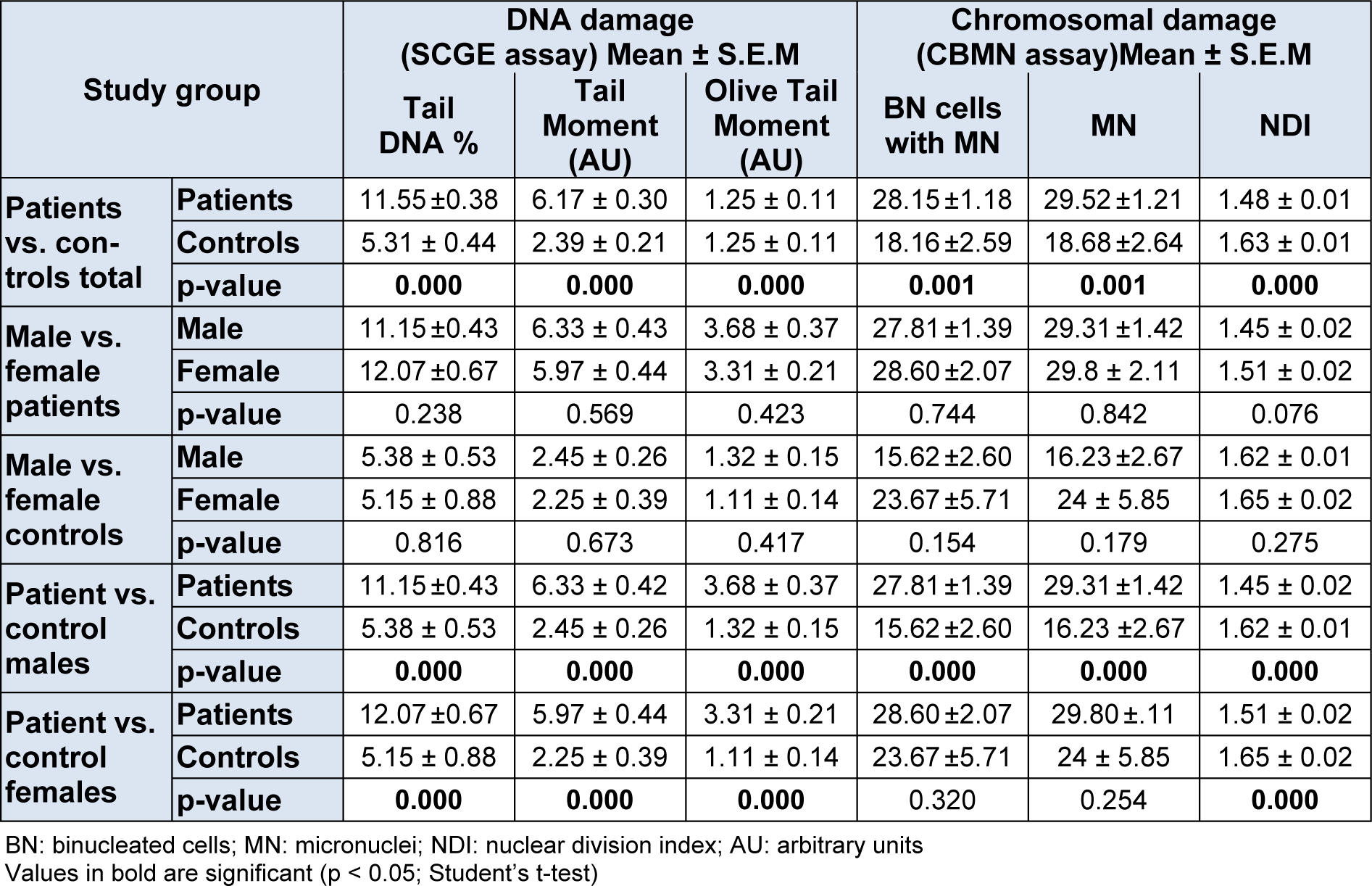
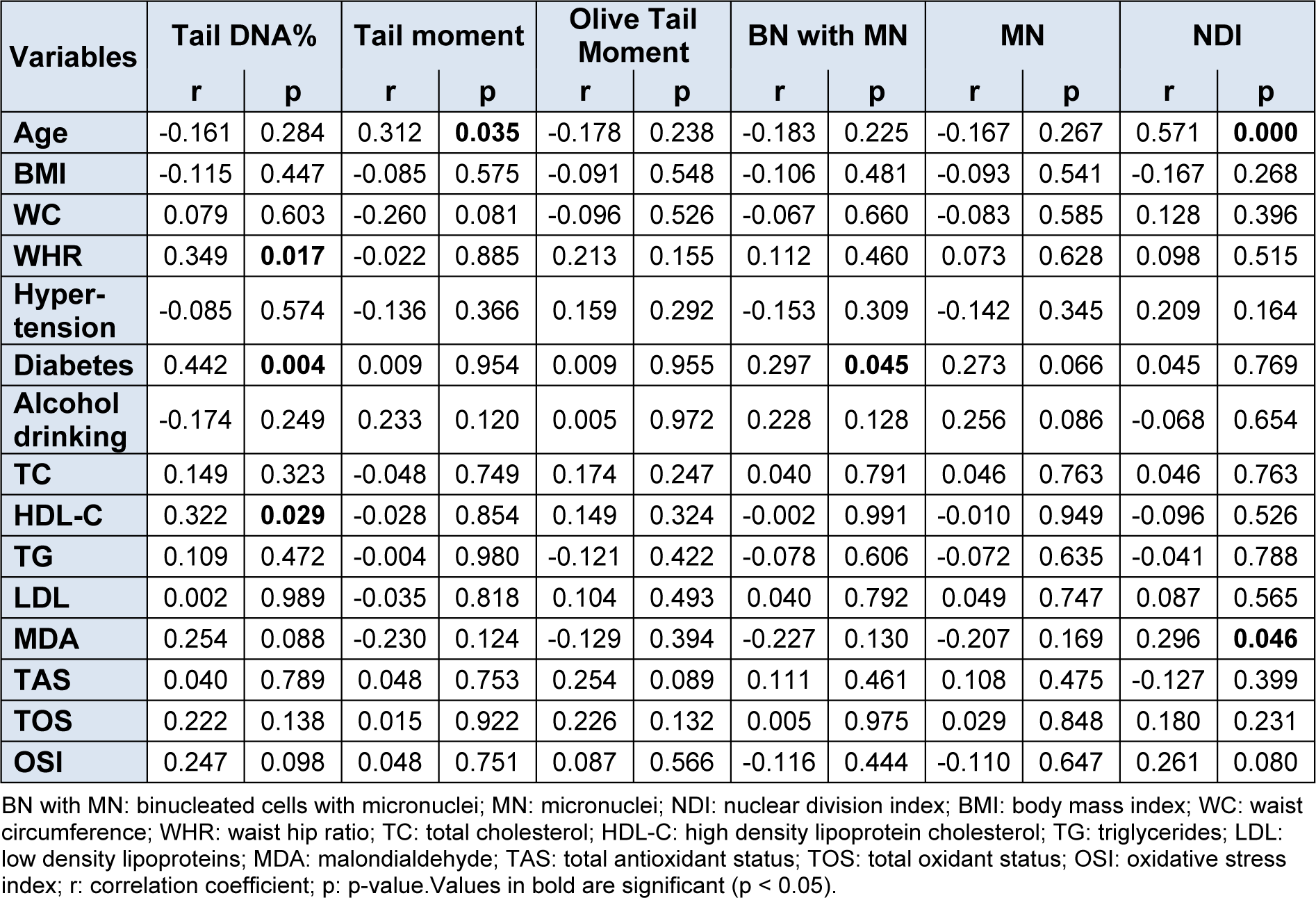
Table 3: Bivariate correlation analysis of DNA and chromosomal damage parameters with demographic and clinical factors
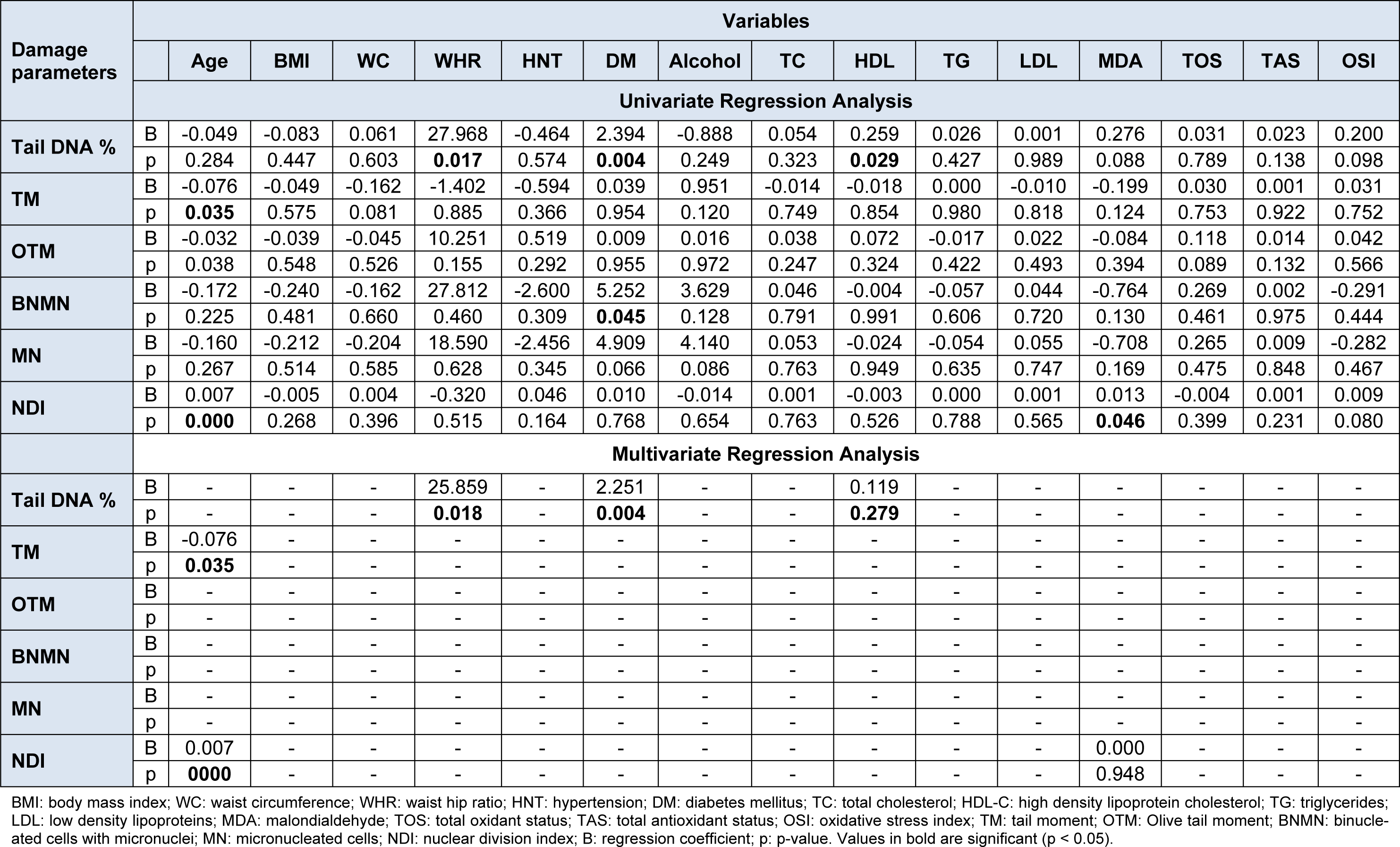
Table 4: Univariate and multivariate regression analyses: DNA and chromosomal damage with demographic and clinical factors
[*] Corresponding Author:
Mohd Akbar Bhat, Department of Human Genetics, Guru Nanak Dev University Amritsar 143005, eMail: akbargenetics@gmail.com