Research article
3-Aryl-1-phenyl-1H-pyrazole derivatives as new multitarget directed ligands for the treatment of Alzheimer's disease, with acetylcholinesterase and monoamine oxidase inhibitory properties
Ashwani Kumar1[*], Sandeep Jain1, Milind Parle1, Neelam Jain2, Parvin Kumar3
1Drug Discovery and Research Laboratory, Department of Pharmaceutical Sciences,Guru Jambheshwar University of Science and Technology, Hisar -125 001, Haryana, India2Department of Pharmaceutical Education and Research, BPSMV, Khanpur kalan, Sonepat, Haryana, India
3Department of Chemistry, Kurukshetra University, Kurukshetra-136119, Haryana, India
EXCLI J 2013;12:Doc1030
Abstract
A series of 3-aryl-1-phenyl-1H-pyrazole derivatives was synthesized in good yield and assayed in vitro as inhibitors of the mice acetylcholinesterase (AChE) and two goat liver monoamine oxidase (MAO) isoforms, MAO-A and MAO-B. Most of the compounds demonstrated a good AChE and selective MAO-B inhibitory activities in the nanomolar or low micromolar range. N-((3-(4-chlorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene) benzenamine (3e, pIC50 = 4.2) and N-((4-fluorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene) methanamine (3f, pIC50 = 3.47) were the most potent AChE and highly selective MAO-B inhibitors respectively. Structure activity relationships showed that chloro derivatives were more effective AChE inhibitors as compared to fluoro derivatives while reverse trend was observed in MAO-B inhibitory activity. With the aid of modeling studies, potential binding orientations as well as interactions of the compounds in the AChE and MAO-B active sites were examined.
Keywords: Alzheimer's disease, 1H-pyrazole, AChE, MAO-B, molecular modeling
Introduction
Alzheimer's disease (AD) is a chronic neurodegenerative disorder and one of the most frequent causes of mental impairment in the elderly (Barnes et al., 2003[2]). It is characterized by the growth of senile plaques and neurofibrillar tangles, which are linked with neuronal loss (Cutler and Sramek, 2001[10]). The past two decades have witnessed a considerable research endeavor devoted to unraveling the molecular, biochemical, and cellular mechanisms of AD (Bartolucci et al., 2006[3]). To date, several factors including amyloid-β (Aβ) deposits, oxidative stress, dyshomeostasis of biometals and low levels of acetylcholine (ACh) have been demonstrated to be associated with AD pathogenesis, and several hypotheses based on these factors have been proposed to explain the mechanism of AD development. Thus, new therapeutic advances that target these features of AD pathology and guarantee disease modification are currently under progress.
At present, management of AD patients continues to be primarily symptomatic, with cholinesterase (ChE) inhibitors as first-line remedy (Terry and Buccafusco, 2003[23]; Xie et al., 2013[30]). On the other hand, changes in further neurotransmitter systems, mainly dopaminergic and serotoninergic (Garcıá-Alloza et al., 2005[16]; Terry et al., 2008[24]) are also believed to be responsible for the behavioral disorders found in AD patients (Dringenberg, 2000[12]). This piece of information has led to the clue that monoamine oxidase inhibitors (IMAOs) might also be important for the treatment of AD (Bolea et al., 2011[6]). Consequently, monoamine oxidase (MAO), flavine adenine dinucleotide (FAD) containing enzyme which catalyze the α-carbon oxidation of a range of biogenic and xenobiotic amines (Strydom et al., 2013[22]) is also a key target to be explored for the treatment of particular facets of this multifactorial disease (Bolea et al., 2011[6]).
Certainly, this multifunctional character of Alzheimer's disease (AD) offers the logical basis for the development of an innovative drug design approach centered on multitarget-directed-ligands (MTDLs) (Bolognesi et al., 2009[7]; Cavalli et al. 2008[8]; Youdim and Buccafusco, 2005[31]). Therefore, the multitarget-directed ligand paradigm has been in the focus of growing attention by many research groups, which have developed a diversity of molecules acting on extremely dissimilar targets (Li et al., 2013[18]; Costa et al., 2013[9]; Elsinghorst et al., 2007[15]).
Multipotent ligands able to simultaneously inhibit cholinesterases, as well as MAOs, have been already developed in the context of a MTDL approach, leading to the discovery of HLA20, M30 and ladostigil, a brain selective molecule acting on different targets on the CNS which is in clinical trials for the treatment of AD (Passos et al., 2013[20]).
Inspired by the particulars discussed above, we report herein design, synthesis, evaluation and molecular modeling studies of compounds as dual inhibitors, active against both acetylcholinesterase as well as monoamine oxidase.
Design of Molecules
It has been proved that derivatives of 1-thiocarbamoyl-3-phenyl-5-thienyl-2-pyrazoline are active in micromolar range against both AChE and MAO-B (Ucar et al., 2005[27]). One interesting compound “a” is shown in Figure 1(Fig. 1).
Now, it has been shown that a high number of sp2 carbons and therefore the π orbitals, increases the molecular recognition by the AChE enzyme as the active site of AChE is composed of many aromatic amino acids (Trujillo-Ferrara et al., 2003[26]).
This idea prompted us to aromatize the 1H-pyrazole ring present in the molecule along with atom bioisosterism and replace the thienyl ring with 6-membered aromatic ring as the latter is present in tacrine and is involved in interactions with enzyme. Also, biological testing of some compounds with imine functionality has shown combined activities both on AChE and MAO. More importantly, the compounds appeared to be reversible MAO inhibitors, potentially devoid of the undesirable side effect associated with the old irreversible inhibitors (Cavalli et al., 2008[8]). So, imine functionality is also introduced into the structure.
Keeping above facts in mind, various 1H-pyrazole derivatives have been designed as shown in Figure 1(Fig. 1).
Results and Discussion
Chemistry
The synthetic path of the 3-aryl-1-phenyl-1H-pyrazole derivatives (2a-3j) is demonstrated in Figure 2(Fig. 2). Straightforward condensation of phenylhydrazine and 4-haloacetophenone gave compounds 1a and 1b which were further cyclized and formylated by means of familiar Vilsmeier-Haack reagent (Youssef et al., 2010[32]) to yield pyrazole carbaldehyde 2a and 2b. The needed products 3a-3g were obtained in good yield by reaction of either 2a or 2b with different amines in the presence of catalytic quantity of glacial acetic acid. Physicochemical characterization of the prepared compounds is given in Table 1(Tab. 1).
Characterization of all the compounds was accomplished by the spectral (IR, NMR) means as well as elemental analysis and found to be in accord with their assigned chemical structures. For example, IR spectrum of the compound 3a revealed characteristic absorption band at 1662 cm-1 due to imine (C=N) group. The pyrazole ring displayed three bands at 1451 cm-1, 1491cm-1, 1111 cm-1 owing to (-C=C),(-C=N), (-N-N) groups respectively. The formation of imine was further confirmed by 1H NMR spectra which exhibited downfield appearance of a singlet at δ 8.72. The pyrazole proton was verified by emergence of one singlet around δ 8.51. The multiplets between δ 7.26-7.86 substantiated the presence of aromatic protons. One singlet at δ 2.75 proved three aliphatic protons.
AChE inhibition evaluation
The inhibitory activities of compounds 2a, 2b and 3a-3j, against AChE (from mice) were measured according to the modified spectrophotometric method of Ellman et al. (Ellman et al., 1961[14]; Vogel, 2002[28]) with donepezil as reference compound.
The AChE inhibition data (IC50 and pIC50 values) of the compounds under study is presented in Table 2(Tab. 2). Interestingly, these results showed that AChE inhibitory activity of the compounds was in nanomolar range and all compounds inhibited the enzyme successfully. Specially, compound 3e and 3j, in which phenyl group was coupled to pyrazole scaffold through imine linker, were the most effective inhibitors with pIC50 value of 4.2 and 4.14 respectively which were comparable to the standard donepezil (pIC50 = 4.92). Inhibition potential of the molecules varied with the substituent on imine nitrogen. There was reduction in activity with increase in the length of the alkyl substituent (3a, pIC50 = 3.90 >3b, pIC50 = 3.82 >3c, pIC50 = 3.75 and 3f, pIC50 = 3.86 >3g, pIC50 = 3.64 >3h, pIC50 = 3.54). Moreover, addition of hydroxyl group enhanced the activity (3b, pIC50 = 3.64 <3d, pIC50 = 3.88). However placing phenyl group at this nitrogen was highly favorable for AChE inhibitory activity (3e and 3j). Overall, chloro derivatives were more potent than fluoro derivatives. Further, introduction of imine functionality augmented the activity as the two aldehydes under investigation were least effective (2a, pIC50 = 3.5, 2b, pIC50 = 3.6) with compound 3h as exception.
MAO inhibition study
To complete the study of the multipotent biological profile of the designed compounds, the inhibitory activity against MAO-A and MAO-B was determined and compared with the inhibition exerted by the standard donepezil.
MAO isoforms of goat liver were used to determine MAO-A and MAO-B inhibitory activities of synthesized pyrazole derivatives using reaction conditions described by Holt et al. (1997[17]) with some modifications. Liver tissue was utilized to screen the MAO-inhibitory actions of these compounds since liver was reported to be a fine source for both isoforms of the enzyme (Bayazit and Khan., 2005[4]).
The MAO-A, MAO-B inhibition data and the selectivity indexes (SI = IC50 MAO-A/IC50 MAO-B) of the present compounds are reported in Table 2(Tab. 2), together with the results obtained for the reference used.
Normally, the newly tested compounds selectively inhibited the enzymatic activity of MAO-B in the nanomolar or low micromolar range. The compounds 2a and 2b were active on both type of isoforms but selective towards MAO-B. Introducing substituted imino functionality in compounds resulted in molecules totally inactive on MAO-A enzyme. Most interesting compounds from the series were 3f (pIC50 = 3.47, SI = >294.12), 3i (pIC50 = 3.43, SI = >263.16) and 3d (pIC50 = 3.22, SI = >166.67). It is noteworthy that lengthening of alkyl chain on imine nitrogen resulted in diminished activity (3a, pIC50 = 3.02 >3b, pIC50 = 2.86 >3c, pIC50 = 2.69 and 3f, pIC50 = 3.47 >3g, pIC50 = 3.24 >3h, pIC50 = 3.14). Further substitution of this alkyl chain with hydroxyl group gave compounds with enhanced activity and selectivity (3b <3d and 3g <3i). The positioning of phenyl group at imine nitrogen produced compounds 3e and 3j. This structural modification caused marked reduction in inhibitory activity. In general, derivatives with p-fluoro group were more effective inhibitors than with p-chloro group. Also, it is notable that transformation of aldehyde to imino was totally unfavourable in case of fluoro derivatives.
Molecular modeling studies
The foregoing studies point out that some compounds seem to be promising multitarget inhibitors. Nevertheless, they also demonstrate unique trends in the pharmacological profile. It is unclear why the inhibitory potency against MAO-B and AChE is affected by the length of the side chain. Therefore, to shed light onto structure activity relationships and to identify the binding mode, a series of docking simulations was conducted for all compounds to AChE and MAO-B. The X-ray crystallographic structures of AChE and MAO-B (PDB code: 1ACJ and 2V5Z) were retrieved from protein data bank. 3D structures of the all AChE inhibitors under study were docked into the active site of the proteins.
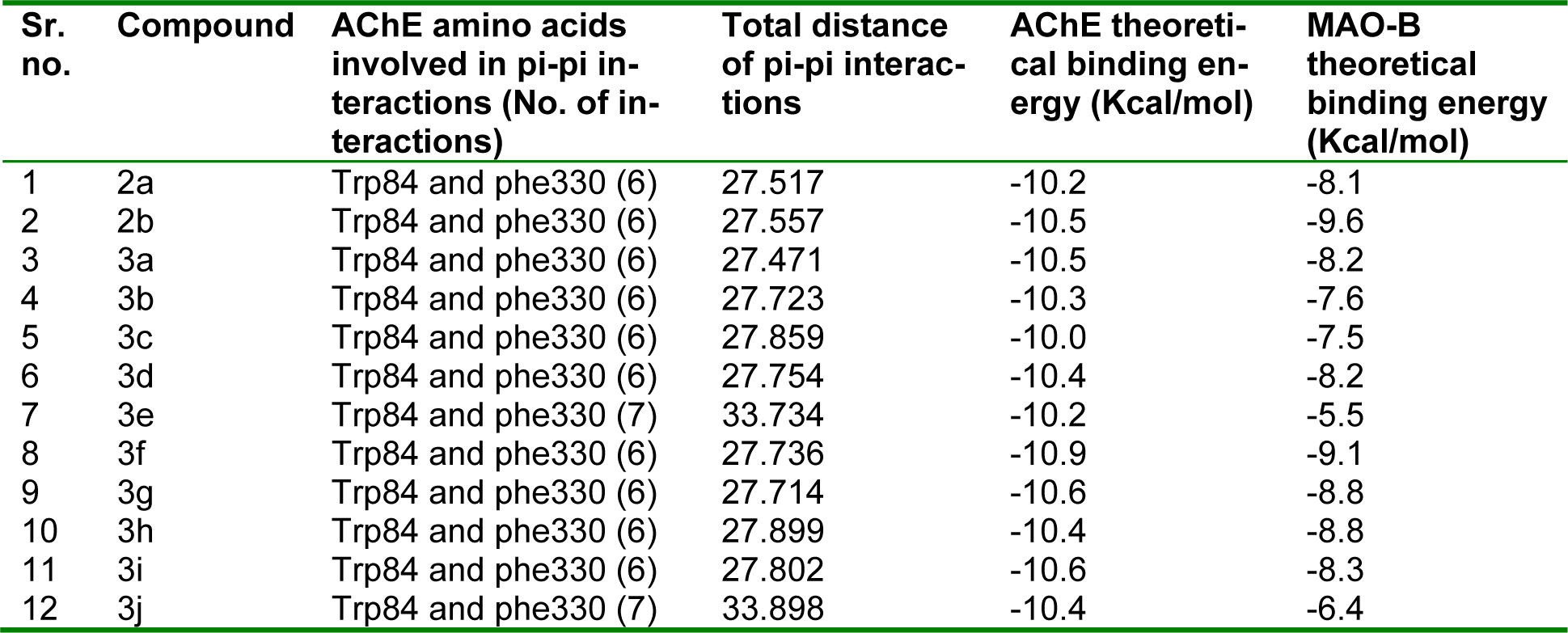
AChE inhibition: All compounds were bound primarily with the anionic subsite of catalytic active site. The ligands were affixed in the active site with pi-pi interactions. Remarkably, all AChE inhibitors were sandwiched between Trp84 and Phe330 by the contribution of aromatic rings. The interaction mode of the top docking pose of most active compound 3e with amino acids in the active site of AChE is depicted in Figure 3(Fig. 3). The theoretical binding energy was not in accordance with the experimental results. However, the experimental activity trend could be explained more or less by the number and the distance of pi-pi stacking interactions. As can be seen from Table 3(Tab. 3) and Figure 3(Fig. 3), compound 3e had highest number of pi-pi stacking interactions and all three aromatic rings were engaged in these. Also, its imine nitrogen was involved in hydrogen bonding with hydroxyl group of TYR121. Although its counterpart 3j possessed same type of binding but the distance of pi-pi interactions was lesser in case of compound 3e than 3j which could be the reason of its higher activity as smaller distance might cause stronger interactions. More activity of all chloro derivatives can be explained on the similar rationale. In case of compounds other than 3e and 3j, simply 1H-pyrazole and 3-phenyl rings were involved in pi-pi interactions which might have been the cause of their reduced activity. Further within both series the activity decreased with increase in interaction distance (except 3d, 3i and 3g). The higher inhibitory potential of 3d and 3i than 3a, 3b and 3f, 3g respectively can be attributed to one extra hydrogen bond formed between hydroxyl hydrogen of both compounds and Gly117 (Figure 4(Fig. 4)). However, the anomalous behaviour of 2a, 2b and 3g could not be put in plain words.
MAO-B inhibition: In this study, theoretical binding affinity was in very good correlation with the observed activity (correlation coefficient = 0.865). Therefore structure activity relationships can be well described using this score. Binding mode of most selective compounds 3f, 3i ad 3d is discussed herein and shown in Figure 5(Fig. 5). The docked compounds exhibited significant contacts with the active site residues. Compound 3f was marvelously accommodated in Aromatic Cage (FAD, TYR398 and TYR435) and its 3-phenyl ring displayed pi-pi interactions with TYR398. Its imine nitrogen formed hydrogen bond with hydroxyl group of TYR435. Nitrogen at 2nd position of pyrazole ring made hydrogen bond with GLN206. Also, fluoro group is involved in hydrogen bonding with FAD. The other two aromatic rings were embedded in Narrow Hydrophobic Cavity comprising of amino acid residues like ILE198 and ILE199. Compound 3i and 3d did not show any pi-pi interactions. Their imine nitrogen established hydrogen bonds with thiol group of CYS172 and hydroxyl group of TYR435. Hydroxyl group of these compounds created contacts with TYR435 and TYR188 through hydrogen bonding. Further these compounds generated hydrogen bond interactions with FAD. The activity of the compounds decreased with increase in the distance from FAD (3f, 3.44 Ao <3i, 3.61Ao <3d, 3.62 Ao).
Experimental Work
All the starting materials and reagents used were of analytical grade and utilized without any further purification. The melting points were determined with Decible Melting Point apparatus and are uncorrected. The reactions were checked by thin layer chromatography (TLC) using silica gel G as stationary phase. Infrared spectra were recorded with Perkin Elmer IR spectrophotometer (KBr disks). Proton nuclear magnetic resonance spectra (1H NMR) were taken on Bruker Avance 400 MHz NMR instrument. Chemical shifts are expressed as δ values (ppm), downfield from tetramethyl silane (TMS) used as internal standard. Splitting patterns are given as s, singlet; d, doublet; t, triplet; m, multiplet.
Chemistry
Procedure for synthesis of phenyl hydrazones (1a, 1b)
To a solution of the appropriate acetophenone (0.04 mol) in 20 mL methanol, phenyl hydrazine (0.04 mol) and glacial acetic acid (1 mL) were added. The resulting solution was shaken for ten minutes at room temperature. The precipitates of hydrazone were filtered and washed with sufficient amount of methanol.
Procedure for synthesis of 3-aryl-1-phenyl-1H-pyrazole-4-carbaldehyde (2a, 2b)
POCl3 (0.06 mol) was added dropwise to an ice-cold stirred solution of hydrazone (1a or 1b) (0.02 mol) in anhydrous DMF (25 mL). Following complete addition of POCl3, the reaction mixture was allowed to attain room temperature and then heated at 60-70 °C for 3-4 h. The resulting mixture was poured onto crushed ice, neutralized with dilute sodium hydroxide. The precipitate obtained were filtered, washed with sufficient quantity of water and recrystalized from chloroform.
3-(4-chorophenyl)-1-phenyl-1H-pyrazole-4-carbaldehyde (2a): IR (KBr, cm-1) ν max: 3042 (Ar-CH), 1664 (C=N),1450, 1493, 1114 (-C=C), (-C=N),(-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 10.02 (s, 1H, -CH=O), 8.51 (s, 1H, Pyrazole-CH), 7.11-7.88 (m, 9H, Ar- H); Anal. Required: C, 67.97; H, 3.92; N, 9.91; Found: C, 67.92; H, 3.95; N, 9.96.
3-(4-fluorophenyl)-1-phenyl-1H-pyrazole-4-carbaldehyde (2b): IR (KBr, cm-1) ν max: 3040 (Ar-CH), 1665 (C=N),1451, 1491, 1111 (-C=C), (-C=N),(-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 10.05 (s, 1H, -CH=O), 8.51 (s, 1H, Pyrazole-CH), 721-7.87 (m, 9H, Ar- H); Anal. Required: C, 72.17; H, 7.14; N, 10.52. Found: C, 72.12; H, 7.16; N, 10.58.
Procedure for synthesis of Schiff bases of 3-aryl-1-phenyl-1H-pyrazole-4-carbaldehyde (3a-3j)
3-Aryl-1-phenyl-1H-pyrazole-4-carbaldehyde (0.02 mol), the appropriate amine (0.04 mol) and catalytic amount of glacial acetic acid were taken in anhydrous methanol and stirred for 72 h at room temperature in case of 3a-3c and 3f-3h whereas refluxed for 06 h for others. The resulting precipitates were filtered, re-crystallized from ethanol and dried at room temperature in desiccator.
N-((3-(4-chlorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene) methanamine (3a): IR (KBr, cm-1) ν max: 3041 (Ar-CH), 1662 (C=N),1451, 1491, 1111 (-C=C),(-C=N), (-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 8.72 (s, 1H, -CH=N), 8.51 (s, 1H, Pyrazole-CH), 7.26-7.86 (m, 9H, Ar- H), 2.75 (s, 3 H, =NCH3); Anal. Required: C, 69.01; H, 11.93; N, 14.24. Found: C, 69.04; H, 11.91; N, 14.23.
N-((3-(4-chlorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene) ethanamine (3b): IR (KBr, cm-1) ν max: 3037 (Ar-CH),1659 (C=N),1453, 1492, 1110 (-C=C), (-C=N),(-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 8.73 (s, 1H, -CH=N), 8.52 (s, 1H, Pyrazole-CH), 7.24-7.80 (m, 9H, Ar- H), 2.72 (q, 2 H, =NCH2); 1.3 (t, 3 H, CH3); Anal. Required: C, 69.79; H, 5.21; N, 13.56. Found : C, 69.75; H, 5.27; N, 13.53.
N-((3-(4-chlorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene) propan-1-amine (3c): IR (KBr, cm-1) ν max: 3035 (Ar-CH), 1664(C=N),1455, 1491, 1112 (-C=C),(-C=N), (-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 8.68 (s, 1H, -CH=N), 8.51 (s, 1H, Pyrazole-CH), 6.20-7.79 (m, 9H, Ar- H), 2.74 (t, 2 H, =NCH2); 1.2 (m, 2 H, CH2) 0.93 (t, 3 H, CH3); Anal. Required: C, 70.47; H, 5.60; N, 12.98.Found: C, 70.44; H, 5.69; N, 12.92.
N-((3-(4-chlorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene) ethanolamine (3d): IR (KBr, cm-1) ν max: 3037 (Ar-CH), 1663 (C=N),1453, 1492, 1110 (-C=C), (-C=N), (-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 8.70 (s, 1H, -CH=N), 8.52 (s, 1H, Pyrazole-CH), 7.19-7.81 (m, 9H, Ar- H), 3.11- 3.42 (m, 4H, CH2); Anal. Required: C, 66.36; H, 4.95; N, 12.90; Found: C, 66.38; H, 4.95; N, 12.96.
N-((3-(4-chlorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene) benzenamine (3e): IR (KBr, cm-1) ν max: 3034 (Ar-CH), 1663(C=N),1455, 1496, 1111 (-C=C),(-C=N), (-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 8.69 (s, 1H, -CH=N), 8.50 (s, 1H, Pyrazole-CH), 7.19-7.82 (m, 14H, Ar- H); Anal. Required: C, 73.84; H, 4.51; N, 11.74 Found:C, 73.81; H, 4.55; N, 11.78.
N-((4-fluorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene ) methanamine (3f): IR (KBr, cm-1) ν max: 3042 (Ar-CH), 1661 (C=N),1453, 1489, 1113 (-C=C), (-C=N),(-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 8.71 (s, 1H, -CH=N), 8.53 (s, 1H, Pyrazole-CH), 7.22-7.85 (m, 9H, Ar- H), 2.75 (s, 3 H, =NCH3); Anal. Required: 73.10; H, 5.05; N, 15.04. Found: C, 73.12; H, 5.07; N, 15.02.
N-((3-(4-fluorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene) ethanamine (3g): IR (KBr, cm-1) ν max: 3040 (Ar-CH),1659 (C=N),1451, 1495, 1112 (-C=C), (-C=N),(-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 8.73 (s, 1H, -CH=N), 8.51 (s, 1H, Pyrazole-CH), 7.21-7.80 (m, 9H, Ar- H), 2.73 (q, 2 H, =NCH2); 1.3 (t, 3 H, CH3); Anal. Required: C, 73.70; H, 5.50; N, 14.32;Found: C, 73.72; H, 5.55; N, 14.30.
N-((3-(4-fluorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene) propan-1-amine (3h): IR (KBr, cm-1) ν max: 3041 (Ar-CH), 1660(C=N),1458, 1490, 1110 (-C=C),(-C=N), (-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 8.67 (s, 1H, -CH=N), 8.50 (s, 1H, Pyrazole-CH), 7.21-7.82 (m, 9H, Ar- H), 2.75 (t, 2 H, =NCH2); 1.2 (m, 2 H, CH2)0.95 (t, 3 H, CH3); Anal. Required: C, 74.25; H, 5.90; N, 13.67. Found: C,74.23; H, 5.91; N, 13.61.
N-((3-(4-fluorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene) ethanolamine (3i): IR (KBr, cm-1) ν max: 3035 (Ar-CH),1660 (C=N),1450, 1491, 1114 (-C=C), (-C=N),(-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 8.70 (s, 1H, -CH=N), 8.52 (s, 1H, Pyrazole-CH), 7.21-7.79 (m, 9H, Ar- H), 3.12- 3.40 (m, 4H, CH2); Anal. Required: C, 69.89; H, 5.21; N, 13.58; Found: C, 69.84; H, 5.22; N, 13.55.
N-((3-(4-fluorophenyl)-1-phenyl-1H-pyrazole-4-yl) methylene) benzenamine (3j): IR (KBr, cm-1) ν max: 3034 (Ar-CH), 1663(C=N),1455, 1496, 1111 (-C=C),(-C=N), (-N-N) pyrazole; 1H NMR (400 MHz, CDCl3, δ ppm): 8.69 (s, 1H, -CH=N), 8.50 (s, 1H, Pyrazole-CH), 7.19-7.82 (m, 14H, Ar- H); Anal. Required: C, 77.40; H, 4.72; N, 12.31; Found: C, 77.42; H, 4.71; N, 12.37.
Biochemistry
Acetylcholinestrase inhibition assay
Spectrophotometric Ellman's method was used for determination of inhibitory activities (IC50 values) of synthesized derivatives 2a-3j against mice AChE using donepezil as reference compound.
Adult male Swiss albino mice (25±5 g) were procured from the Disease Free Small Animal House, Lala Lajpat Rai University of Veterinary & Animal Sciences (LLRUVAS), Hisar (Haryana). The experimental protocol for animal studies was consented by Institutional Animal Ethics Committee (IAEC). The animals were housed in the animal house facility of the Department of Pharmaceutical Sciences, G.J.U S & T, Hisar, under standard conditions of temperature (25±2 °C) and 12 hr/12 hr light/dark cycles. The animals were kept under laboratory conditions for one week before beginning of the experiments and permitted food and water ad libitum.
Male Swiss albino mice were decapitated; brain was rapidly dissected on ice, weighed and homogenized in 19 volumes (about 7 mg protein/ml) of 0.05 M NaH2PO4, pH 7.2. To 4 mL of the vehicle or different concentrations of the test compound, 100 μL aliquot of this suspension was added and reincubated for 10 min at 37 °C.
A stock solution of the test compound (2 mM in dimethylsulfoxide) was made and diluted to required volume using 0.5 mM DTNB reagent. Sequential dilutions (1:10) of the compounds were prepared so as to attain the final concentration of 10-4 M and checked for activity. If active, IC50 values were determined from the inhibitory activity of succeeding concentrations.
Blank, control and test samples were incubated for 150 s with acetylthiocholine iodide (10 mM) and production of the yellow anion of 5-thio-2-nitrobenzoic acid was measured with Systronics 108 UV-visible spectrophotometer at 412 nm.
The percentage inhibition was calculated by the following formula: %inhibition = Absorbance of control-Absorbance of inhibitor/Absorbance of control x 100. Response-logarithm of inhibitor concentration curves were plotted for all compounds and the IC50 values were determined with BioToolKit320 (BioToolKit320, 2005[5]). Results are expressed as the mean ± SD of at least three different experiments.
Measurement of MAO activity
The synthesized compounds were also screened for their activity at MAO-B and MAO-A using in vitro techniques.
MAO was purified from the goat liver (100-150 g). Liver samples of goat were purchased from local market immediately after sacrifice. Homogenates of liver were prepared with 1:25 (w/v) in ice-cold potassium phosphate buffer, pH 7.4 with a mechanical homogenizer. Homogenates were centrifuged at 1000 g, at 4 °C for 15 min and the supernatant was used as the source of MAO and kept at -70 °C. Since, MAO-A activity decreased rapidly following homogenization, freshly prepared homogenates were used in studies of MAO-A activity (Bayazit and Khan, 2005[4]).
MAO activity was evaluated spectrophotometrically according to the method of Holt et al. (1997[17]). Homogenates were incubated with the substrate p-tyramine (500 µM to measure MAO-A and 2.5 mM to measure MAO-B) following the inhibition of one of the MAO isoforms with selective inhibitors. Aqueous solutions of clorgyline or pargyline (500 nM), as selective MAO-A and -B inhibitor, were added to homogenates. Homogenates were incubated with these inhibitors at 37 °C for 60 min earlier to activity determination. After incubation with test compounds (10 nM - 100 µM) or control, the MAO reactions were started and the reactions were incubated at 37 °C. The assay mixture contained a 150 µL chromogenic solution (1 mM vanillic acid, 500 µM 4-aminoantipyrine, and 4 U mL-1 peroxidase in 0.2 M potassium phosphate buffer, pH 7.6) 600 µL substrate solution (500 µM p-tyramine), and 150 µL potassium phosphate buffer, pH 7.6. The mixture was preincubated at 37 °C for 10 min before the addition of enzyme. Reaction was initiated by adding the homogenate (100 µL), and absorbance was monitored at 498 nm at 37 °C after 60 min. The results were expressed as IC50 and pIC50 values.
Molecular modeling
The 2D structures of compounds were built with MavinSketch (MavinSketch 5.10.1., 2012[19]) and explicit hydrogens were added. Afterward these structures were optimized, changed into 3D and cleaned with gradient optimization. The resulting structures were saved in pdb format. The X-ray crystallographic structures of AChE along with tacrine and MAO-B with inhibitor Safinamide (PDB ID: 1ACJ and 2V5Z) were obtained from Brookhaven Protein Databank (http://www.rcsb.org/pdb). Molecular modeling software UCSF Chimera (Pettersen et al., 2004[21]) was employed for preparation of protein for docking. All solvent molecules and co-crystallized ligands except FAD (2V5Z) were removed and imperfect side chains were completed using Dunbrack rotamer library (Dunbrack, 2002[13]). After that hydrogens were added and Gasteiger charges were computed with ANTECHAMBER (Wang et al., 2006[29]). The prepared molecules were saved in pdb files for further workout. These structures of ligands and proteins were changed into pdbqt format by means of AutoDock tools (AutoDock Tools 1.5.6 rc2[1]).
Docking simulations were performed with AutoDock Vina (Trott and Olson, 2010[25]) program. The Vina search space applied was center_x = 4.34518462891, center_y = 69.9038811926, center_z = 65.7756741596, size_x = 25.0, size_y = 25.0, size_z = 25.0 for 1ACJ and center_x = 52.1640844198 center_y = 155.977828543 center_z = 27.8383407001 size_x = 22.5898271723 size_y = 25.0 size_z = 4.3118371451 for 2V5Z. The exhaustiveness was set to be 8.
Validation of the docking protocols was done with reported crystal structures of protein-ligand complexes. The root-mean square deviation (RMSD) amongst the conformations of the Tacrine and Safinamide from the X-ray crystal structure and those from the results of AutoDock Vina was less than 1 Å, recommending that the parameters chosen for the AutoDock Vina simulation were practical to imitate the X-ray structures. These docking protocols were employed for docking of the compounds under investigation into the binding pocket of target enzymes. Final results were visualized with the help of Discovery Studio (Discovery Studio v3.5 client, 2012[11]).
Conclusion
In the present study, a new family of multitarget molecules able to interact with AChE as well as MAO-B has been synthesized and evaluated. Moreover, a brief idea regarding their structure activity relationship was drawn and significance of different substitutions predominantly at imine nitrogen was studied. Presence of any substitution at imine nitrogen improved the AChE inhibitory activity and altered the selectivity in the direction of MAO-B. The binding mode analysis of compounds with the assistance of molecular docking simulations bestowed imperative insights about their molecular recognition process. The data of this research suggests these molecules as promising leads for the development of novel MTDL with a good AChE and MAO-B inhibitory potency, which are presently missing in the therapeutic arsenal.
References
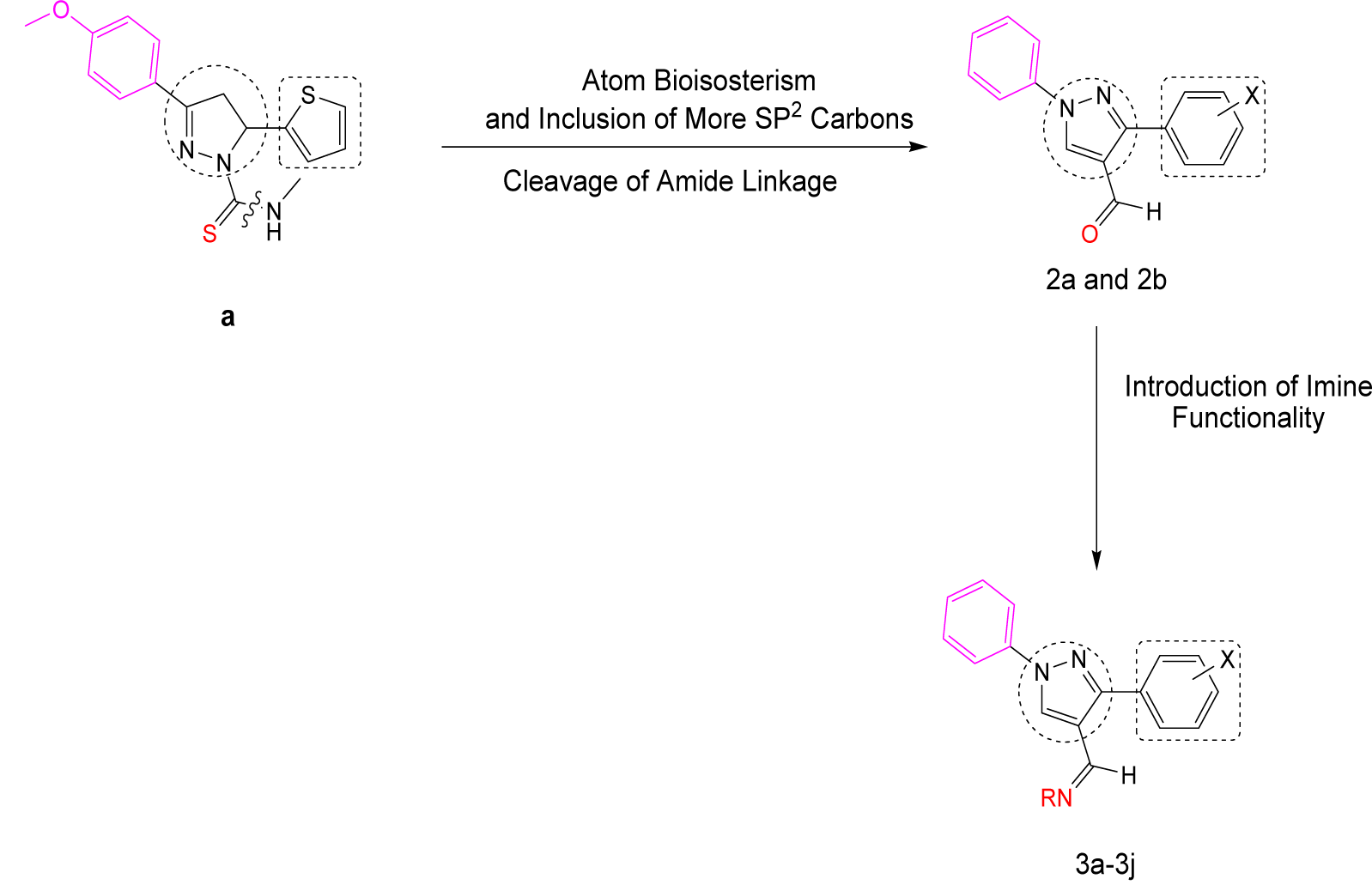
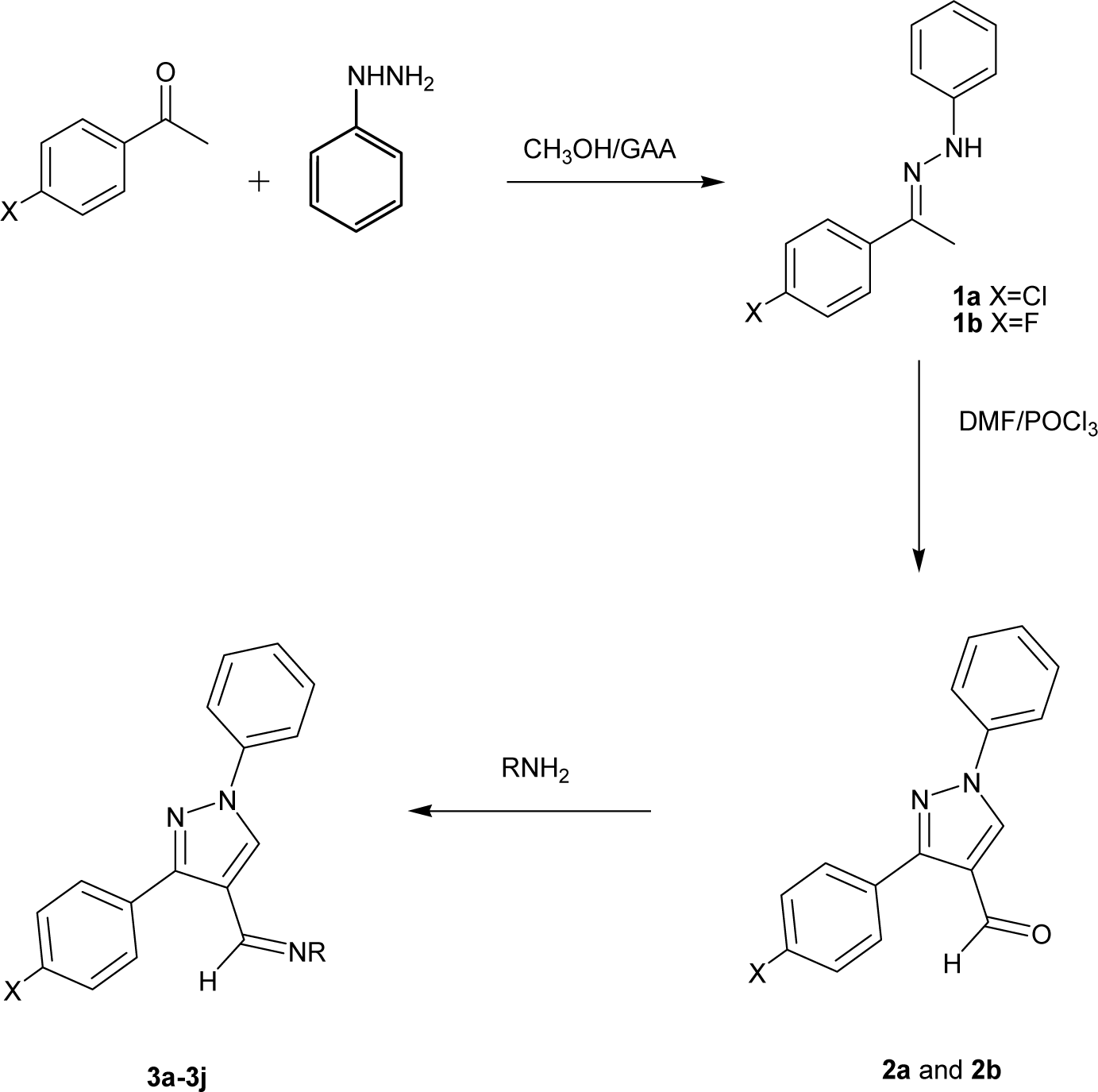
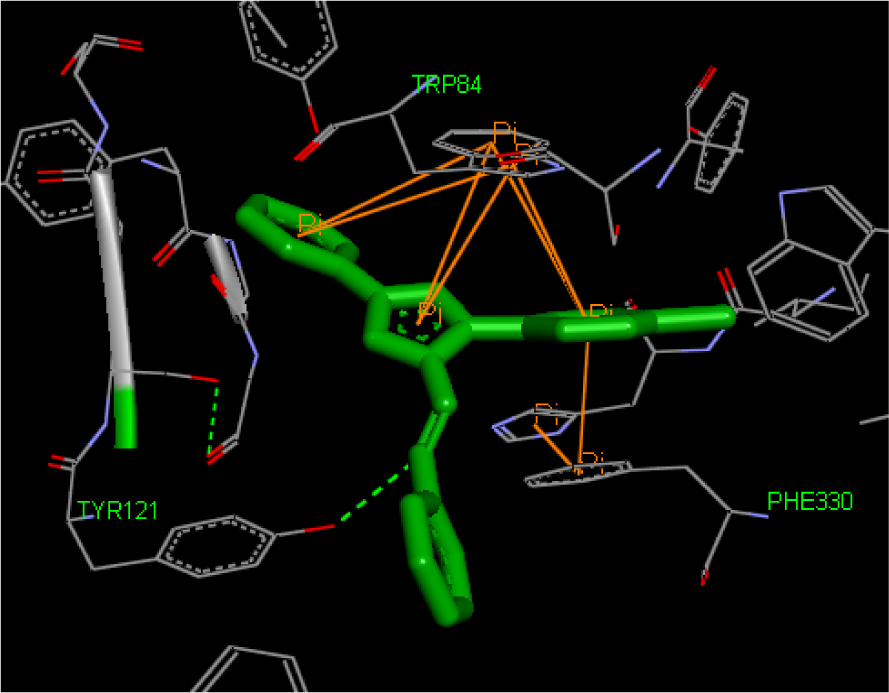
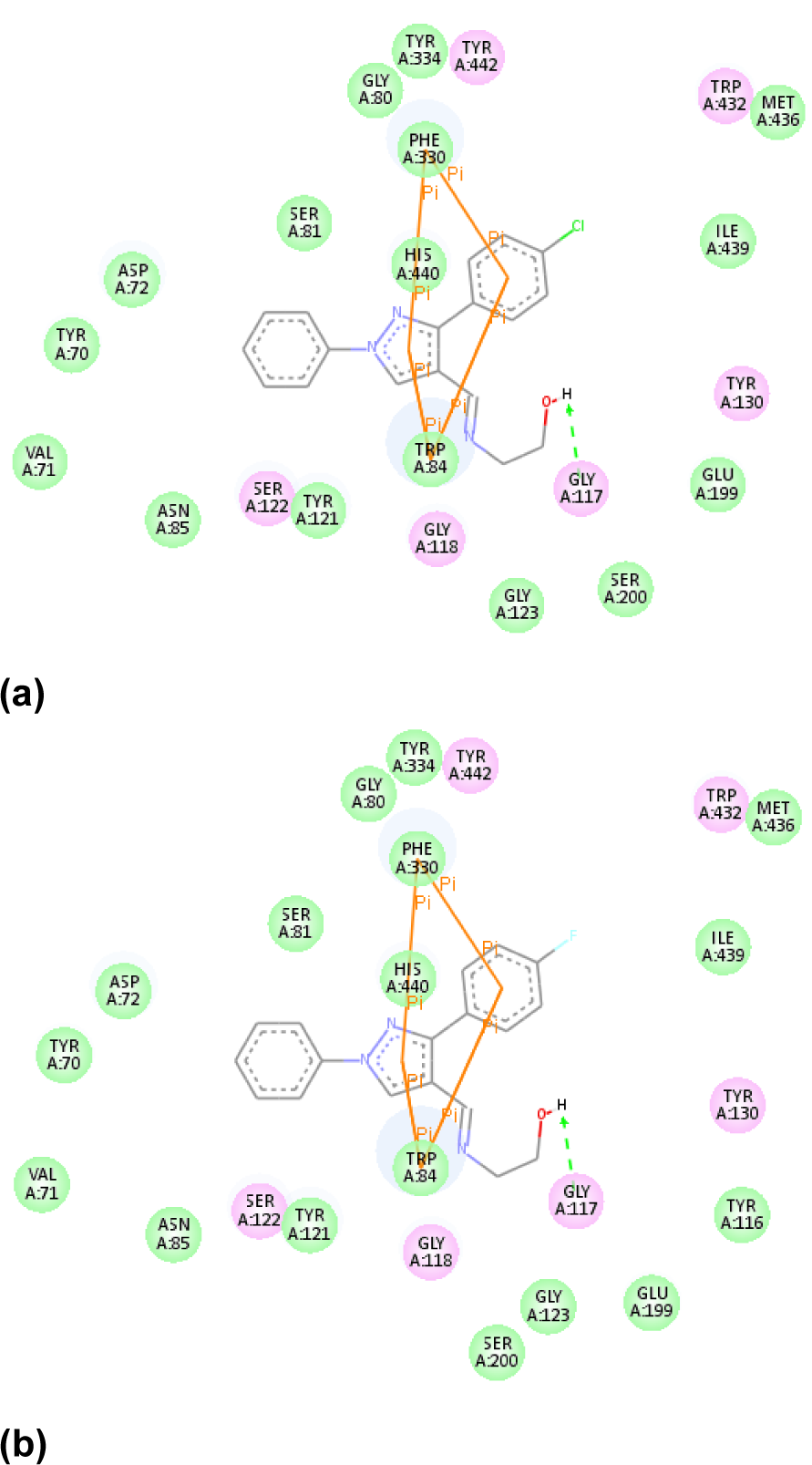
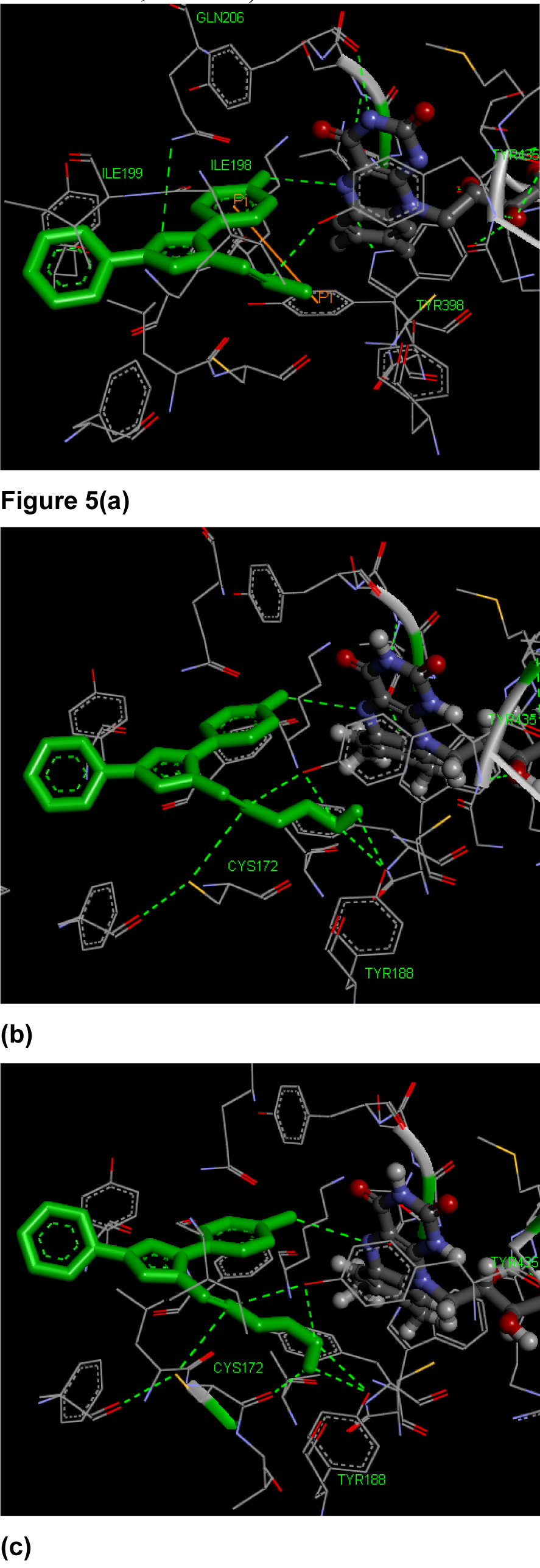
Figure 5: Interactions of compounds (a) 3f (b) 3i and (c) 3d with MAO-B active site containing FAD (ball and stick model)
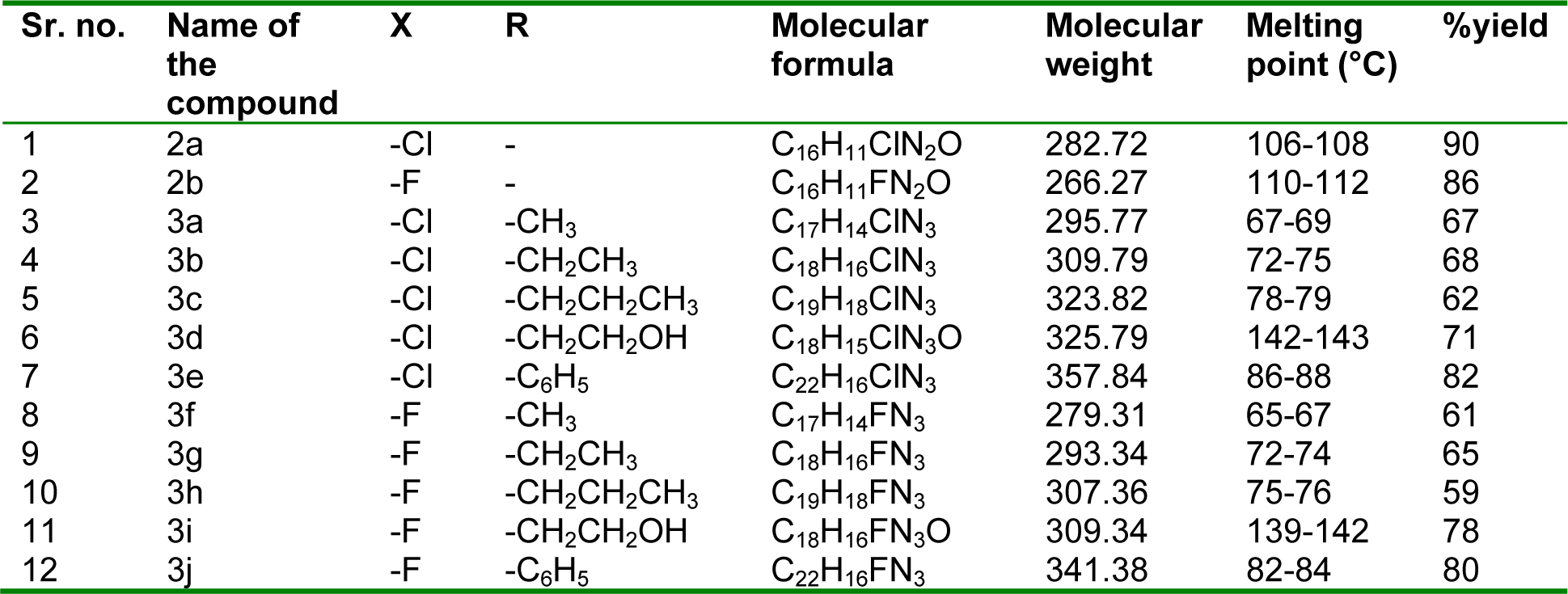
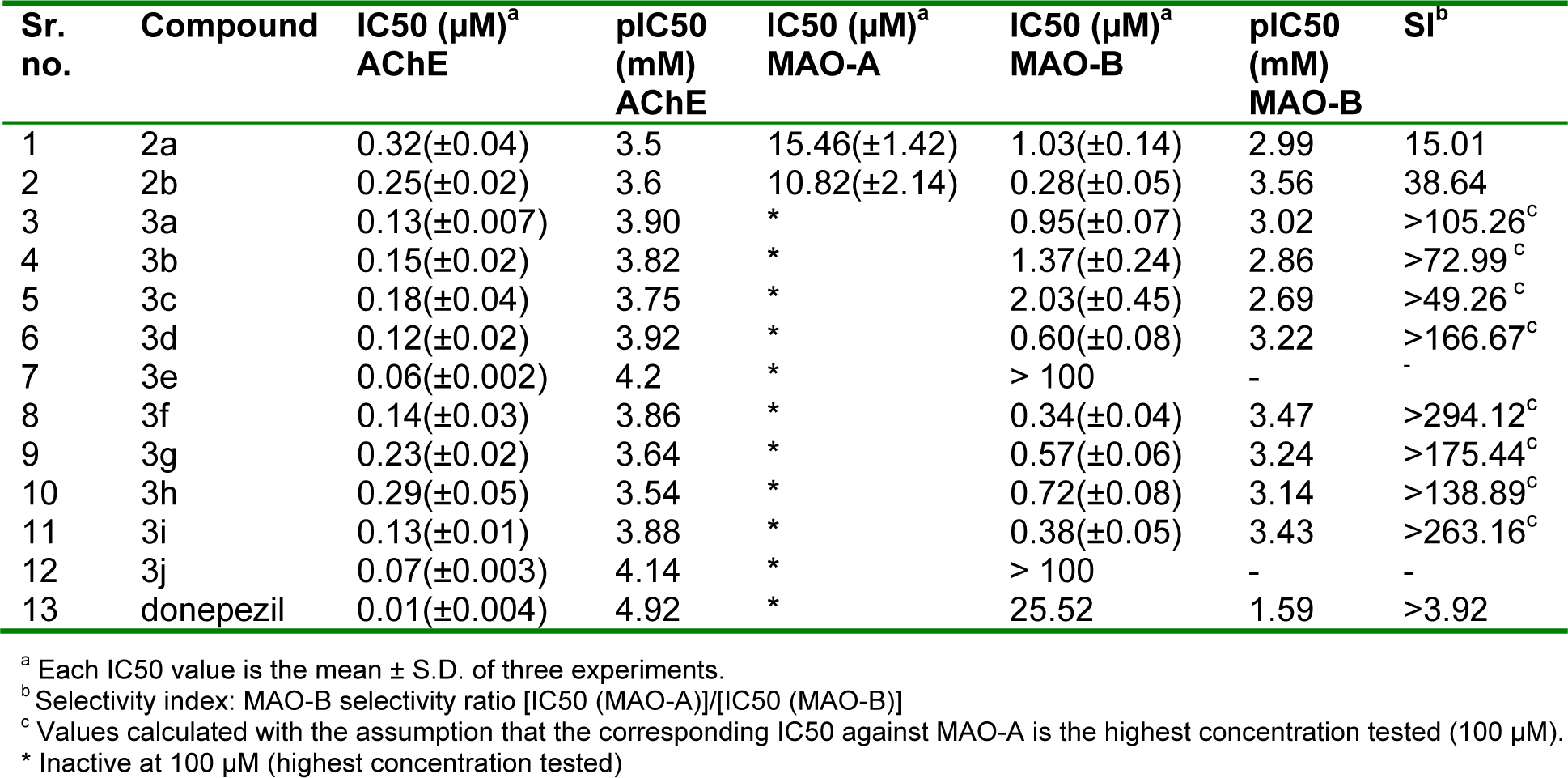
Table 2: In vitro AChE, MAO-A and MAO-B inhibitory activities of 1H-pyrazole derivatives and reference compound
[*] Corresponding Author:
Assistant Prof. Ashwani Kumar, Drug Discovery and Research Laboratory, Department of Pharmaceutical Sciences, Guru Jambheshwar University of Science and Technology, Hisar -125001, INDIA; Tel.: 09466280487; Fax: +91-1662-276240, eMail: ashwanijangra@ymail.com