Review article
miRNA regulation during cardiac development and remodeling in cardiomyopathy
K.L. Chaitra1, Kayalvili Ulaganathan1, Anita James1, Venkateshwari Ananthapur2, Pratibha Nallari1[*]
1Department of Genetics, Osmania University, Hyderabad, India2Institute of Genetics and Hospital for Genetic Diseases, Hyderabad, India
EXCLI J 2013;12:Doc980
Abstract
miRNAs have been found to play a major role in cardiomyopathy, a heart muscle disorder characterized by cardiac dysfunction. Several miRNAs including those involved in heart development are found to be dysregulated in cardiomyopathy. These miRNAs act either directly or indirectly by controlling the genes involved in normal development and functioning of the heart. Indirectly it also targets modifier genes and genes involved in signaling pathways. In this review, miRNAs involved in heart development, including dysregulation of miRNA which regulate various genes, modifiers and notch signaling pathway genes leading to cardiomyopathy are discussed. A study of these miRNAs would give an insight into the mechanisms involved in the processes of heart development and disease. Apart from this, information gathered from these studies would also generate suitable therapeutic targets in the form of antagomirs which are chemically engineered oligonucleotides used for silencing miRNAs.
Keywords: miRNA, cardiomyopathy, modifiers, signaling pathways
Introduction
Cardiomyopathy is a disease of the heart muscle characterized by cardiac dysfunction, arrhythmia, heart failure and sudden death. It is a major cause of morbidity and mortality worldwide. Cardiomyopathy can be classified into 2 major groups based on predominant organ involvement: Primary cardiomyopathy (genetic, nongenetic, acquired) are those which are confined to heart muscle and Secondary cardiomyopathy show pathological myocardial involvement as part of a large number and variety of generalized systemic disorders (Elliott et al., 2008[16]; Maron et al., 2006[31]).
It is also considered as a disease of sarcomere and cytoskeleton and recent studies have revealed new facets about the role of micro RNAs (miRNAs) in cardiac development and remodeling. A growing body of exciting evidence suggests that miRNAs are regulators of cardiovascular cell differentiation, growth, proliferation, angiogenesis and apoptosis. It has provided glimpses of undiscovered regulatory mechanisms and potential therapeutic targets for the treatment of cardiomyopathy.
microRNAs
miRNA, an endogenous ~22nt RNA, plays an important regulatory role in animals and plants. They cleave the target mRNA or translationally repress the mRNA. The first miRNA discovered was lin-4 in C. elegans, which was found to produce a pair of short RNA transcripts that regulate the timing of larval development by translational repression of lin-14, which encodes a nuclear protein (Lee et al., 1993[29]). Since the discovery of let-7, miRNAs have been identified in organisms as diverse as viruses, worms, and primates through random cloning and sequencing or computational prediction. A hepta nucleotide sequence at the 5' end of the miRNA is essential in the base pairing specificity with the 3' end of the mRNA (Gregory et al., 2008[19]).
Role of miRNA in Heart Development and Function
Role of miRNAs in heart development was identified in zebrafish and mice by targeting the Dicer protein which is the main component of miRNA machinery. Zebrafish lacking maternal and endogenous dicer exhibited heart developmental defects (Giraldez et al., 2005[18]; Wienholds et al., 2003[58]). Cardiac-specific deletion of dicer in mice resulted in pericardial edema and poorly developed ventricular myocardium, resulting in embryonic lethality (Zhao et al., 2007[62]). Adult mice lacking dicer in the myocardium revealed high incidence of sudden death due to cardiac hypertrophy and reactivation of the fetal cardiac gene program (da Costa Martins et al., 2008[13]). Further studies also showed that additional miRNAs, miR1, miR133, miR1/133 (bicistronic), miR21 and miR138 are involved in the regulation of heart development.
miR-1/miR-133: The most abundant miRNA in cardiac myocytes and also the first miRNA implicated in heart development was miR-1(Zhao et al., 2007[62]). miR-1 and -133 are expressed in cardiac and skeletal muscles and are transcriptionally regulated by the myogenic differentiation factors MyoD, Mef2, and serum response factor (SRF) (Chen et al., 2008[9], Kwon et al., 2005[27]; Lagos-Quintana et al., 2001[28]; Rao et al., 2006[36]; Sokol and Ambros, 2005[47]; Zhao et al., 2005[63]).
Targeted deletion of the muscle-specific miRNA, miR-1-2, was found to cause ventricular septal defects (VSD) in embryos of mice resulting in immediate death and/or pericardial edema which contributes to embryonic mortality (Yu et al., 2008[61]). In mice that survived postnatally, some died within months due to rapid dilatation of the heart and ventricular dysfunction, while many others suffered sudden cardiac death because of abnormalities in cardiac conduction and repolarization. miR-1-2 also directly targets irx5, which is known to repress the potassium channel, Kcnd2, ensuring coordinated cardiac repolarization (Costantini et al., 2005[12]). In adult miR-1-2 mutants, cardiomyocyte division continues postnatally due to abnormalities in cell-cycle leading to hyperplasia of the heart.
miR-1 levels are low during cardiac development but seem to increase as development progresses. In mice, overexpression of miR-1 under the control of the myosin heavy chain (MHC) promoter negatively regulates cardiac growth, by inhibiting translation of heart and neural crest derivative-2 protein, Hand2, which is involved in ventricular myocyte expansion (Zhao et al., 2005[63]). In drosophila, dmiR-1 plays an important role in differentiation of cardiac progenitor cells by targeting transcripts of delta, a ligand involved in Notch signaling pathway, which regulates the expansion of cardiac and muscle progenitor cells (Kwon et al., 2005[27]).
miR1/133 (bicistronic) has been found to enhance differentiation of mouse and human embryonic stem cells into mesoderm while suppressing their differentiation into neuroectoderm or endoderm. But later during the adaptation of muscle lineage, miR-1 and miR-133 had opposing effects, wherein miR-1 promotes differentiation into cardiac muscle fate and miR-133 blocks the cell fate (Ivey et al., 2008[22]). Stress induced apoptosis is provoked by increased levels of miR-1, and increased levels of miR-133 has a protective effect against cell death triggered by free radicals such as hydrogen peroxide (Tang et al., 2009[48]; Xu et al., 2007[59]; Cheng et al., 2007[10]).
miR-21: Another miRNA consistently induced by cardiac stress is miR-21, the role of which is vaguely defined. This particular miRNA is stress induced and is a regulator of cardiac growth (Van Rooij et al., 2006[53]). Hence its interaction with the HSP-70 gene family needs to be elucidated, as HSP is also a cardiac specific development gene. Sabatel et al. (2011[39]) showed that over-expression of miR-21 reduced endothelial cell proliferation, migration, and tube formation by targeting RhoB, whereas knockdown of miR-21 led to an opposite effect.
miR-138: miR-138 is a highly conserved miRNA, which is found in many regions of the developing embryo, but within the zebrafish heart, it is categorically expressed in the ventricular chamber (Morton et al., 2008[32]). Changes in miR-138 function resulted in the expansion of the atrioventricular canal into the ventricle and failure of maturation of ventricular cardiomyocytes. It has thus been suggested that other region-specific miRNAs may reinforce known signaling and transcriptional networks that establish patterns of gene expression throughout the developing heart tube (Cordes and Srivastava 2009[11]). Hence the relation between modifier genes and miRs during embryogenesis needs to be focused in the current research on cardiomyopathy.
miRNA in Cardiomyopathy and Cardiac Remodelling
miRNA mediated gene repression is an important regulatory mechanism to modulate fundamental cellular processes such as the cell cycle, growth, proliferation, phenotype, death, which in turn may have major influence on the pathophysiological outcomes like cardiac fibrosis, hypertrophy, angiogenesis, and heart failure (Catalucci et al., 2009[8]; Thum et al., 2007[51], 2008[52]). Although miRNAs are highly expressed in heart, their role in heart diseases especially cardiomyopathy still remains elusive. Multiple aberrant miRNA expression is a remarkable characteristic of the hypertrophic heart (Cheng et al., 2007[10]). The dysregulation and the time course changes of these multiple aberrantly expressed miRNAs match the complex process of cardiac hypertrophy formation in which several genes have been reported to be dysregulated (Dorn and Hahn, 2004[14]). Determining the effects of these dysregulated miRNAs in cardiac hypertrophy is the prerequisite for novel research on cardiomyopathy. It was reported that a cardiac-specific knockout of the Dicer gene leads to rapidly progressive dilated cardiomyopathy (DCM), heart failure, and postnatal lethality. Dicer expression decreased in end-stage DCM heart due to heart failure. These findings suggest that Dicer function and miRNAs play critical roles in normal cardiac function and heart diseases especially during heart failure (Chen et al., 2008[9]).
miR-1/miR-133: miR-133 and miR-1 play critical roles in cardiac hypertrophy. In human and mouse models of cardiac hypertrophy, decreased expression of both miR-133 and miR-1 is reported. In vitro, overexpression of miR-133 or miR-1 inhibited cardiac hypertrophy. In contrast, suppression of miR-133 induced hypertrophy. In vivo inhibition of miR-133 by a single infusion of an antagomir caused marked and sustained cardiac hypertrophy. RhoA, a GDP-GTP exchange protein (regulator of cardiac hypertrophy); Cdc42, a signal transduction kinase (implicated in hypertrophy); and Nelf-A/WHSC2, a nuclear factor (involved in cardiogenesis) are all targets of miR-133 (Carè et al., 2007[7]).
When overexpressed in normal or infarcted rat hearts, miR-1 aggravates arrhythmogenesis and elimination of miR-1 by an antisense inhibitor relieved arrhythmogenesis. The target of miR-1 was found to be gap junction protein α1 (GJA1) which encodes Cx43, the main cardiac gap junction channel important for conductance in the ventricles (Luo et al., 2007[30]) and potassium inwardly-rectifying channel, subfamily J, member 2 (KCNJ2) (Yang et al., 2007[60]).
miR-21: Inhibition of miR-21 in neonatal rat cardiomyocytes by transfecting locked nucleic acid (LNA)-modified antisense oligonucleotides (miR-LNA) to miR-21 or miR-18b induced myocyte hypertrophy while transfection of miR-21 and miR-18b duplexes slightly decreased cardiomyocyte size and decreased hypertrophy, thus suggesting their role in regulating cardiomyocyte hypertrophy. The molecular basis of such regulation still needs to be established. Hence the role of miR-21 in chromatin modelling of cardiomyocytes cannot be overlooked (Tatsuguchi et al., 2007[49]).
A study showed an increased expression of miR-21, miR-23, miR-24, miR-125b, miR-195, miR-199a, and miR-214, and decreased expression of miR-29c, miR-93, miR-150, and miR-181b in mice subjected to chemicals which induced cardiac hypertrophy. Upon transfection into cardiomyocytes, five of the miRNAs that were upregulated, viz. miR-23a, miR-23b, miR-24, miR-195, and miR-214, were found to be capable of inducing hypertrophic growth. Whereas transfection of miR-199a resulted in elongated spindle shapes myocytes reminiscent of the elongated cardiac myocytes observed in dilated cardiomyopathy (DCM).
Cardiac specific overexpression of miR-195 was found to cause death in the first 2 weeks after birth due to heart failure associated with cardiac dilatation and in the rats that survived initially, induction of cardiac growth with disorganization of cardiomyocytes occurred at 2 weeks of age, which later progressed to a dilated cardiac hypertrophic phenotype by 6 weeks of age thus suggesting the critical role played by miR-195 in cardiac remodeling (Van Rooij et al., 2006[53]).
miR-29: The miR-29 family, which is downregulated after myocardial infarction, inhibits expression of several collagens and extracellular matrix proteins, thereby contributing to scar formation and fibrosis, as seen in DCM, during heart failure.
miR-208: miR-208 is required for the development of cardiac hypertrophy and myocardial fibrosis and it is also a positive regulator of MHC gene expression (van Rooij et al., 2007[54]).
Regulation of Modifiers by miRNAs
Stress is a major etiologic factor that may contribute to heart diseases. Stress overload can cause tissue injury and cardiomyocyte death is considered an important cellular basis for stress-induced injury in cardiomyopathy (Feuerstein and Young 2000[17]). miR-199 family is rapidly downregulated in cardiac myocytes under hypoxic conditions, relieving the repression of sirtuin 1 and hypoxia inducible factor 1-α in a model of hypoxia preconditioning. The miRNA that repeatedly showed dynamic regulation after cellular stress is miR-21, which promotes cardiac hypertrophy and fibrosis in response to pressure overload (Rane et al., 2009[35]).
Under stress, a change in expression of HSP70 in rat myocardium is observed. HSP70 protects cardiomyocyte from stress induced injury by inhibiting Fas-mediated apoptosis (Basu et al., 2001[4]). Levels of miR-1 were found to increase significantly in response to oxidative stress which later reduced the levels of HSP70 favoring cardiomyocyte apoptosis, while decreased levels of miR-1 favored cardiomyocyte survival (Xu et al., 2007[59]). Members of TGF-β family also have been found to have a cardioprotective role and are highly induced in affected hearts. Their putative roles during atherogenesis, infarct healing, cardiac repair and left ventricular remodeling have been proposed (Os et al., 2002[34]). miR24a and miR34a seem to have a strong and specific regulatory effect on TGF β while miR-373 and miR-34b have a constitutive role (Schultz et al., 2011[43]). The renin-angiotensin system (RAS) is one of the most important modifiers in cardiomyopathy. The angiotensin converting enzyme (ACE) is the key enzyme, involved in conversion of angiotensin I to angiotensin II which is responsible for cardiac hypertrophy and heart failure (Kawaguchi, 2003[24]) and miR145 has been found to regulate this enzyme (Small et al., 2010[46]). Thus it can be deliberated that diseased phenotype is influenced by either cytoskeletal/sarcomeric genes, modifiers, and/or miRNAs controlling these genes.
Hence it can be hypothesized that miRNAs have both positive and negative roles in cardiomyopathy, especially with respect to a heart failure phenotype; either as modifiers or by gene-gene interaction and/or regulators of cardiogenesis. One such regulation could be in the signaling/transduction pathways controlled by the miR's.
Regulation of Signaling Pathways by miRNAs and their Role in Cardiomyopathies
Understanding complex diseases like cardiomyopathy not only requires identification of genes and upregulation/ downregulation of miRNAs, but also of the proteins that are regulated and signaling pathways that are affected by these miRNAs. Various intercellular signaling pathways have been implicated in the control of cardiogenesis viz. Notch signaling, FGF signaling, BMP signaling, Wnt/β-catenin signaling, Wnt/ JNK pathways etc.
(I) Notch Signaling and Cardiogenesis
Notch signaling mediates numerous developmental cell fate decisions in organisms ranging from flies to humans, resulting in the generation of multiple cell types from equipotential precursors. Notch signaling is also involved in angiogenesis and vasculogenesis. Notch signaling is a highly conserved and a complex mechanism initiated by the interaction of Notch receptors with their ligands both of which are transmembrane proteins whose extracellular domains are composed of epidermal growth factor (EGF) like repeats (Eiraku et al., 2005[15]). The Notch receptors include the Notch1-4 in mammals and Notch in drosophila. The Notch ligands include classical ligands such as Jagged - Jag1 and Jag2 - and Delta-like - DLL1, DLL3 and DLL4 and atypical ligands DNER, F3/Contactin1, NB-3/Contactin6 and Delta-like 1 homologue. The Notch pathway is intricately involved in the development of the cardiovascular system. One of the major functions of Notch signaling is its ability to influence cell fate decisions during development (Kwon et al., 2005[27]). Several of the Notch pathway components have been linked to the vascular system development, including Jagged1, Notch1, Notch2, Notch4 and presenilin (Eiraku et al., 2005[15]; Bray et al., 2008[5]). The Notch1 receptor is responsible for the blockade of cardiogenesis. Notch1 is also involved in the suppression of cardiomyocyte differentiation. It has also been proposed that inhibition of cardiogenesis by Notch signaling is carried out by blocking mesodermal differentiation (Sethupathy et al., 2006[44]). Hence Notch signaling pathway, known to influence cardiogenesis and heart development, in conjunction with miRNAs, needs to be elucidated.
Notch Signaling and miRNA in Cardiomyopathy
miRNA regulation is essential for normal Notch signaling. Default repression by miRNAs does not necessarily have to target core pathway components; it may be equally effective when it intercepts their transcriptional targets as shown by the default repression of the E (spl) and Bearded (Brd) gene clusters whose activation is dependent on signaling by Notch in Drosophila. This is a highly redundant system, in which families of related miRNAs (miR-2, miR-4, miR-7, miR-11 and miR-79) promiscuously target a family of related mRNAs, preventing aberrant deployment of Notch-mediated developmental programmes (Sabatel et al., 2011[39]). Regulation of the expansion of cardiac and muscle progenitor cells is carried out by the notch ligand Delta, and this is targeted for repression by dmiR-1 (Kwon et al., 2005[27]). Several conserved putative miR-1-binding sites were found in the 3'-UTR of the gene encoding Delta (Artavanis-Tsakonas et al., 1999[1]). It was also found that miR-1 fine-tunes Notch ligand Delta that is critically involved in differentiation of cardiac and somatic muscle progenitors and targets a pathway essential for progenitor cell specification and asymmetric cell division. Introduction of miR-133 allows cardiac tissue formation, but the tissue is disorganized and does not lead to chamber formation. It has thus been shown that miR-1 and miR-133 function antagonistically to each other whenever miR-1 shifts the development of the stem cells towards a cardiac fate and miR-133 inhibits this event. The cardiac fate achieved by miR-1 is by transcriptional repression of Dll-1, which is the mammalian ortholog of Delta in Drosophila (Atsuhiko et al., 2011[2]). It has also been reported that the members of the Hairy family, particularly Hrt2/Hey2, involved in heart disease, are themselves regulated by miR-1-2 and members of the Hairy family are transcriptional repressors which mediate Notch signaling. The effect produced by miR-1-2 on Hey2 is also seen on Hand1, involved in Notch pathway, which is a bHLH transcription factor involved in ventricular development and septation that, in combination with Hand2 (a paralog of Hand1), is known to regulate expansion of the embryonic cardiac ventricles (Kwon et al., 2005[27]; Jiang et al., 2009[23]; Rusconi and Corbin, 1998[38]; Sabatel et al., 2011[39]; Sapir et al., 2005[41]). miR-1-2 appears to be involved in the regulation of diverse cardiac and skeletal muscle functions, including cellular proliferation, differentiation, cardiomyocyte hypertrophy, cardiac conduction and arrhythmias (Han and Bodmer, 2003[20]). Hence studying miRs regulating the Notch signaling pathway involved in cardiac development, differentiation and ultimately cardiomyopathy needs to be emphasized.
(II) Wnt/β-catenin and Wnt/JNK Signaling in Cardiogenesis
Wnt proteins are secreted glycoproteins which upon binding to Frizzled family receptors and several coreceptors such as lipoprotein receptor-related protein (LRP)-5/6, Ryk, or Ror2, activates intracellular signal transduction. Some aspects of cell cycle, differentiation and proliferation are dependent on regulation of gene expression by Wnt proteins. Cell adhesion requires the interaction of β-catenin with cadherins by establishing a link to the actin cytoskeleton (Rao and Kühl, 2010[37]). During early embryonic development of zebrafish, the Wnt/ β-catenin pathway induces the formation of the lateral mesoderm, whereas its negative role/repressive activity helps to define the proper size of the heart-forming field (Tazhor, 2007[50]).
Depending on the major intracellular mediators, Wnt/jun N-terminal kinase (JNK) or Wnt/calcium pathways, which are independent of β-catenin are activated. Activation of small GTPases of the rho family including rac, cdc42, and rho and other downstream protein kinases such as JNK or rho kinase is involved in the Wnt/JNK pathway. Dkk1, a target gene of Wnt/β-catenin signaling establishes a negative-feedback loop by way of inhibiting the canonical Wnt pathway. The N-terminal domain of Dkk1 may be involved in regulating cardiogenesis, in a β-catenin independent fashion (Korol et al., 2008[25]).
miRNA and Wnt Signaling
β-catenin dependent transcription is down regulated by miR-200a by two different mechanisms. MiR-200a induces cell-cell adhesion by increasing the level of E-cadherin, via targeting the mRNA of the E-cadherin repressor proteins ZEB1 and ZEB2 (Korpal et al., 2008[26]). Following which, β-catenin is marked for degradation by ubiquitin/proteasome system through phosphorylation at Ser33, Ser 37, Thr 41 and Ser45 (Burk et al., 2008[6]). Another mechanism of downregulation of β-catenin activity by miR-200a was recently proposed in meningiomas. MiR-200a regulates β-catenin expression and subsequent activation of Wnt/ β-catenin signaling by directly targeting the 3'UTR of β-catenin mRNA. Other members of miR-200 family, viz. miR-200b and miR-200c have no effect in this domain (Saydam et al., 2009[42]).
MiR-135a and miR-135b stabilize β-catenin and thus activate Wnt signaling by suppressing APC expression (Nagel et al., 2008[33]). MiR-315 directly targets Axin and Notum, two negative regulators of Wg signaling, in Drosophila cells, resulting in Wnt pathway activation (Silver et al., 2007[45]; Huang K et al., 2010[21]).
Thus it can be said that MiRNAs indirectly regulate cardiogenesis by way of β-catenin in WNT signaling.
(III) FGF and BMP Signaling in Cardiogenesis
Fibroblast Growth Factor (FGF) and Bone Morphogenetic Proteins (BMP's) are cardiogenic inducers. FGFs are involved in the induction and patterning of the mesoderm and their role in cardiogenic induction involves synergistic interaction with BMP signaling pathways. The need for both BMP and FGF in cardiac induction was revealed in experiments showing that BMP-2 alone could not promote survival of precardiac or non-precardiac mesoderm cells in culture, whereas FGF-4 could support and maintain cardiogenesis in precardiac mesoderm, although it lacked the ability to induce cardiogenesis in non-precardiac mesoderm (Wagner and Siddiqui, 2007[55]).
While BMP and FGF pathways are implicated in promoting cardiac induction, there is evidence that Wnt antagonism stimulates cardiogenesis in chick and Xenopus embryos. In apparent contrast, Wnt/β-catenin signaling promotes cardiogenesis in ES cells if activated early, but opposes it when activated after specification (Samuel and Latinkic, 2009[40]).
miRNA in FGF and BMP Signaling
BMP-2 increases Runx2 expression which results in increased expression of the Runx2-dependent genes osteopontin and osteocalcin, increased intracellular Ca2+ deposition, and calcification of human coronary artery smooth muscle cells (CASMCs). BMP-2 downregulates miR-30b and miR-30c to increase Runx2 protein expression and vascular calcification. In C2C12 cells, BMP-2 promotes osteogenic differentiation by decreasing miR-133 ex-pression to increase Runx2 as well as downregulating miR-206 expression to limit the expression of myogenic differentiation marker proteins. BMP-2 was also shown to regulate Runx2 expression by upregulating miR-3960 expression which resulted in decreased expression of homeobox A2, a repressor of Runx2 expression (Balderman et al., 2012[3]).
BMP signaling regulates the miRNA-17-92 complex through Smad-mediated pri-miRNA transcriptional activation to control a pathway promoting SHF myocardial differentiation. Other important cardiovascular miRNAs, such as miRNA-143 and miRNA-145, are downregulated in BMP 2/4 CKO mutants but some other miRNAs are upregulated suggesting that BMP signaling likely regulates multiple events in cardiac development through miRNA effector pathways.
Bmp2 and Bmp4 regulate OFT (outflow tract) myocardial differentiation via regulation of the miRNA-17-92 cluster. In BMP mutant embryos, myocardial differentiation was delayed, and multiple miRNAs encoded by miRNA-17-92 were reduced. Genetic interaction studies uncovered a synergistic interaction between miRNA-17-92 cluster and BMP 4, providing direct in vivo evidence for the BMP-miRNA-17-92 regulatory pathway. These findings indicate that BMP signaling directly regulates a miRNA-mediated effector mechanism via downregulation of cardiac progenitor genes and enhancement of myocardial differentiation (Wang et al., 2010[56]).
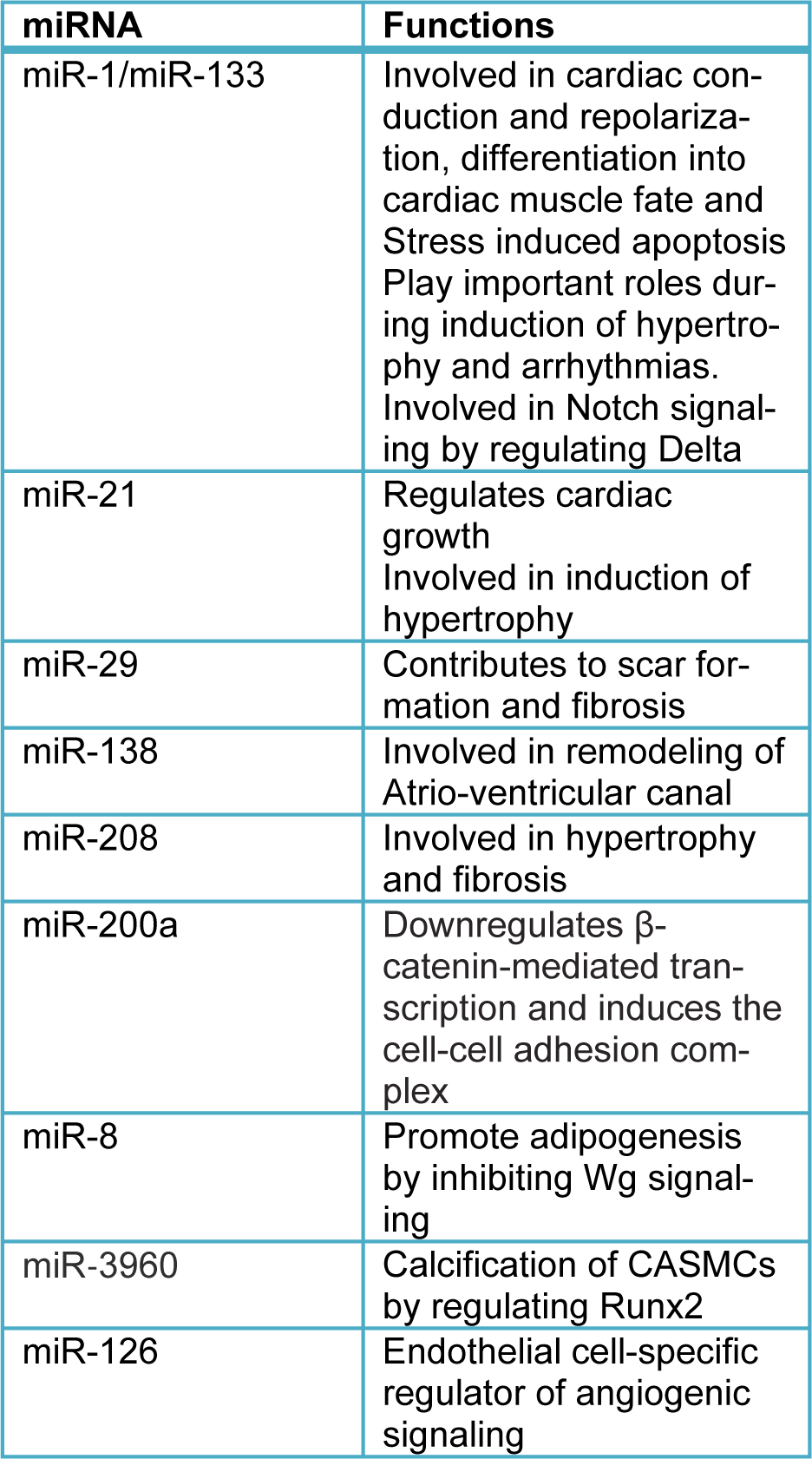
Potent angiogenic growth factors, such as VEGF and FGF, are required for neoangiogenesis during embryogenesis and adulthood. miR-126 which is an endothelial cell-specific miRNA, modulates angiogenesis in vivo, but in a subset of mutant mice, targeted deletion of miR-126 resulted in vascular leakage, hemorrhaging, and embryonic lethality, and these aftereffects can be attributed to reduced angiogenic growth factor signaling. The proangiogenic action of miR-126 correlates with its repression of Spred-1, a negative regulator of MAP kinase signaling. Hence decline in the transmission of intracellular angiogenic signals by VEGF and FGF culminated because of increased expression of Spred-1 in the absence of miR-126. Hence it can be concluded that miR-126 functions as an endothelial cell-specific regulator of angiogenic signaling. The vascular abnormalities of miR-126 mutant mice simulate the outcome of decreased signaling by angiogenic growth factors, such as VEGF and FGF. miR-126 augments the proangiogenic actions of VEGF and FGF and promotes blood vessel formation by repressing the expression of Spred-1, an intracellular inhibitor of angiogenic signaling. These findings will help in improvising the current therapeutics for a variety of disorders involving abnormal angiogenesis and vascular leakage. miR-126 is required for vascular integrity and angiogenesis, as well as survival post-Myocardial Infarction and its potential for augmenting cardiac repair needs to be utilized. Diminishing miR-126 expression may be efficacious in settings of pathological vascularization, such as cancer, atherosclerosis, retinopathy and stroke. Thus strategies to promote miR-126 in the ischemic myocardium which could intensify cardiac repair need to be devised. Hence the exact role of miR-126 and associated miRNAs in cardiac repair needs to be elucidated to understand their functioning in tissue remodeling and diseases (Wang et al., 2008[57]). A brief summary of the miRNAs with their various functions is given in Table 1(Tab. 1).
Conclusion
In the preceding discussion, the involvement of miRNAs in regulating developmental processes in the heart and their involvement in cardiomyopathy via sarcomeric genes, modifiers and signaling pathways such as the Notch pathway is reviewed. Mutations in sarcomeric genes are the primary causatives of the disease, whereas the modifiers determine the severity.
The miRNAs regulating these genes thus play an important role in development and disease. The roles played by several miRNAs have been elucidated, but an in depth analysis of the miRNAs, and the genes that encode them and also the genes targeted by them is essential to bring forward the complex interplay that occurs during development and disease causation. Notch pathway is involved in the development of cardiovascular system, as it promotes cell proliferation and apoptosis. Many miRNAs are known to regulate the Notch pathway and the dysregulation of these miRNA affects cell proliferation, differentiation, cardiac conduction, leading to cardiac hypertrophy and arrhythmias. But the information available in this context is still obscure. Further studies are necessary to identify other miRNAs involved in regulation of notch pathway. The effect of miRNA regulation through WNT signaling pathway is evident only through its control of β-catenin, other dimensions are yet to be discovered. The BMP-FGF pathways is important in subsidiary processes of cardiogenesis such as myocardial differentiation and angiogenesis, indicating the role of miRNA in the regulation of these pathways. A study of miRNAs would also give us potential therapeutic targets in the form of antagomirs which are used for silencing miRNAs that are implicated in the manifestation of cardiomyopathy. Complete revelation of the roles played by miRNA may give crucial insights into many of the mysteries of the human heart and CVD's and taking into view the benefits of these miRNA's and associated pathways, miRNA-based therapies may pave the way for the treatment of cardiomyopathy.
Notes
K.L. Chaitra, Kayalvili Ulaganathan and Anita James share first authorship.
References
[*] Corresponding Author:
Head and Professor, Dr. Pratibha Nallari, Department of Genetics, Osmania University, Hyderabad, India, eMail: prathinallari@yahoo.com