Research article
Macrophage migration inhibitory factor is involved in endovascular trophoblast cell function in vitro
Aleksandra Vilotic1, Milica Jovanovic Krivokuca1[*], Ivana Stefanoska1, Svetlana Vrzic Petronijevic2, Miloš Petronijevic2, Ljiljana Vicovac1
1Laboratory for Biology of Reproduction, Institute for the Application of Nuclear Energy, INEP, University of Belgrade, Banatska 31b, 11080 Belgrade, Serbia2Clinic of Obstetrics and Gynecology, Clinical Center of Serbia, Koste Todorovica 26, 11000 Belgrade, Serbia
EXCLI J 2019;18:Doc1007
Abstract
Macrophage migration inhibitory factor (MIF) is a multifunctional cytokine abundantly present at the feto-maternal interface proposed to play a role in establishment of pregnancy. We have previously shown that pharmacological inhibition of enzymatic activity of MIF decreases extravillous trophoblast invasion and migration in vitro. This study aimed to further elucidate potential role of endogenous trophoblast MIF, and to assess its importance for endovascular trophoblast cell function in particular. Attenuation of MIF by siRNA reduced HTR-8/SVneo cell invasion through Matrigel (59 % of control), expression of integrin α1 (86 % of control) and levels of MMP2 and MMP9 (87 % and 57 % of control, respectively). MIF specific siRNA reduced the ability of HTR-8/SVneo to differentiate in to endothelial-like phenotype, as determined by Matrigel tube formation assay. The total tube length was decreased to 68.6 %, while the number of branching points was reduced to 57.8 % of control. HTR-8/SVneo cell capacity to integrate into HUVEC monolayers was reduced by knock-down of MIF. This could be partly caused by reduced N-cadherin expression to 63 % of control, which decreased with knock-down of MIF, as the expression of this protein was recently shown essential for trophoblast-endothelial interaction. These novel findings indicate a novel role for trophoblast MIF in spiral artery remodeling process.
Keywords: trophoblast, HTR-8/SVneo, HUVEC, N-cadherin
Introduction
Embryo implantation in mammals with hemochorial placenta involves multiple critical steps, such as embryo attachment to the uterine epithelium, translocation in to the uterine stroma and subsequent invasion of local tissues by extravillous trophoblast (EVT) lineage formed along the process. During the first trimester of human pregnancy interstitial EVT invade decidual stroma and part of myometrium anchoring placenta to the mother's uterus while endovascular EVT invade decidual spiral arteries transforming their vessel walls by degrading and replacing endothelial and smooth muscle cell layers (Pijnenborg, 1998[28]). Remodeled spiral arteries become dilated high-flow conduits escaping vasomotor control thus enabling constant supply of maternal blood to the feto-maternal interface which is important for normal development of the growing fetus (Pijnenborg, 1998[28]).
During endovascular invasion EVT acquires endothelial-like phenotype which facilitates trophoblast integration and subsequent replacement of the endothelial cells of the spiral arteries. Adhesive molecule repertoire of endovascular EVT changes allowing direct cell-cell interaction between trophoblast and endothelial cells (Burrows et al, 1994[9]; Multhaup et al., 2018[27]; Zhou, et al., 1997[39]). Spiral arteries remodeling includes endothelial cell apoptosis and extracellular matrix catabolism (Ashton et al., 2005[5]; Harris et al., 2010[14]).
Reduced spiral artery remodeling is associated with serious pregnancy pathologies such as pre-eclampsia and fetal growth restriction (Kaufmann et al., 2003[21]; Pijnenborg et al., 2011[30]; Zhou et al., 1997[38]). Although several mechanisms were proposed endovascular invasion and spiral artery remodeling are still not fully understood. Further research on molecules involved and underlying mechanisms of these processes is expected to lead to improved understanding of both physiological and pathological events, which could aid in early detection of the aberrant blood vessel transformation.
MIF is a multifunctional cytokine highly expressed at the feto-maternal interface during pregnancy (Arcuri et al., 1999[3], 2001[4]). The highest level of MIF in human placenta is determined in early pregnancy (Ietta et al., 2007[17]) predominantly expressed in villous cytotrophoblast and extravillous trophoblast (Arcuri et al., 1999[3]). At the end of the first trimester MIF in placenta is decreasing and is maintained to the term (Ietta et al., 2007[17]). Although available data on MIF in human pregnancy imply important role of this cytokine for EVT function and placentation, specific contribution of MIF still remains to be elucidated. Our group previously showed that MIF supports EVT invasion and migration in vitro, while pharmacological inhibition of MIF abolished these processes (Jovanović Krivokuća et al., 2015[19]). MIF protects trophoblast cells from excessive apoptosis (Ietta et al., 2018[16]) and is involved in immune privilege at the feto-maternal interface (Arcuri et al., 2006[2]) and placental response to infection (De Oliveira Gomes et al., 2011[12]; Jovanović-Krivokuća et al., 2016[20]).
This study aimed to further elucidate relevance of endogenous trophoblast MIF for early pregnancy events, more specifically endovascular differentiation of EVT and its potential contribution to trophoblast-endothelial interaction, important for spiral artery remodeling.
Material and Methods
Cell culture
The HTR-8/SVneo (donated by Dr Charles H Graham) (Graham et al., 1993[13]) were maintained in RPMI 1640 medium (Gibco, UK) supplemented with 10 % heat inactivated fetal calf serum (v/v) (FCS, Sigma-Aldrich, USA) and 1 % antibiotic/antimycotic solution (Capricorn Scientific, Germany) (HTR medium). HUVEC were isolated from umbilical cords (5 individual cases) collected after term deliveries (approved by the Ethical committee of the Clinical Centre of Serbia, approval no. 57/10) by sequential short trypsinization as previously described (Jiménez et al, 2013[18]). Collected cells were seeded on 0.2 % gelatin-coated plates and maintained in M199 medium (Lonza, Belgium) supplemented with 20 % heat inactivated FCS, 2 mM L-Glutamine (Torlak, Serbia), 0.4 % endothelial cell growth supplement containing heparin (ECGS/H) (PromoCell GmbH, Germany) and 1 % antibiotic/antimycotic solution (HUVEC medium).
MIF siRNA transfection
Predesigned specific siRNA for MIF (Silencer Select Validated siRNA, s8780) and negative control (Silencer Negative Control siRNA #2, AM4613) were purchased from Ambion (Thermo Fisher Scientific Inc., USA). HTR-8/SVneo cells were transfected with 30 nM siRNA using Lipofectamine RNAiMAX (Invitrogen, USA) according to manufacturer's instruction in antibiotic-free Opti-MEM I GLUTAMAX I Reduced Serum Medium (Opti-MEM) (Gibco, UK). There were 3 experimental groups: L - cells treated with Lipofectamine only, S - cells transfected with negative control siRNA, and siMIF - cells transfected with MIF siRNA. After 48 h or 72 h transfected cells were collected for further analyses.
Cell viability assay
HTR-8/SVneo cell viability was assessed by MTT test 48 h and 72 h following transfection. Cells were collected and seeded in 96-well plates at 5 x 104 cells/well in 100 µl of Opti-MEM medium. 10 µl of MTT (5 mg/ml in PBS) was added to each well and the cells were incubated 3 h at 37 °C, 5 % CO2. At the end of the incubation, 100 µl of 10 % SDS (0.01 N HCl) was added to each well and the plate was further incubated at 37 °C overnight to ensure complete solubilization of formazan. The absorbance was read at 540 nm using a microplate reader (LKB, Austria).
Cell invasion assay
HTR-8/SVneo cells were collected 48 h after transfection and transwell invasion assay was conducted as previously described with minor changes (Stefanoska et al., 2013[33]). Briefly, 1 x 105 cells were seeded on top of Matrigel (Corning, USA)-coated cell culture inserts (8 µm pore size, Merck KGaA, Germany). After 24 h incubation, cells on the upper side of filter inserts were gently removed with cotton swab. After rinsing and fixation, cells were stained by Giemsa, and the occupied pores of the entire filter were counted. All experiments were done in Opti-MEM medium.
Quantitative real-time PCR
qPCR analyses were done as previously described (Bojić-Trbojević et al., 2019[8]). Expression levels of MIF gene (MIF_F: CCGGACAGGGTCTACATCA; MIF_R: ATTTCTCCCCACCAGAAGGT) were normalized to the housekeeping gene GAPDH (GAPDH_F: GAAGGTGAAGGTCGGAGT; GAPDH_R: GAAGATGGTGATGGGATTTC). Calculations were made by the comparative ΔΔCt method (Livak and Schmittgen, 2001[25]).
SDS-PAGE and immunoblot
HTR-8/SVneo whole cell lysates prepared 48 h or 72 h after transfection, or cell conditioned media, were analyzed by Western blot after protein separation on 12.5 % gel as previously described (Bojić-Trbojević et al., 2019[8]) using anti-MIF antibody (0.5 mg/ml, R&D Systems, UK). The obtained signals were scanned and analyzed by the ImageMaster TotalLab v2.01 program (Amersham Biosciences, Inc., USA). MIF protein expression levels were normalized to the expression of GAPDH (anti-GAPDH, 1:12000, Cell Signaling Technology Inc, USA).
Flow cytometry
HTR-8/SVneo cells were collected 48 h or 72 h after transfection by 0.25 % trypsin/0.02 % EDTA. Cells were permeabilized and prepared further for flow cytometry and analyzed as previously described (Bojić-Trbojević et al., 2019[8]) using following primary antibodies: anti-MIF (0.5 µg/ml, R&D Systems, UK), anti-integrin α1 (1 µg/ml, R&D Systems, UK), anti-integrin α5 (1:25; BIIG2), anti-integrin β1 (1:50; AIIB2) (kind gift from Susan Fisher Lab, University of California, San Francisco, USA), and anti-N-cadherin (10 µg/ml, Thermo Fisher Scientific, USA), and anti-mouse Alexa Flour 488 secondary antibody (Molecular Probes, Life Technologies, Thermo Fisher Scientific Inc., USA).
Gelatin zymography
Matrix metalloproteinase (MMP) gelatinolytic activity was determined in conditioned media of HTR-8/SVneo cells collected 48 h after transfection by SDS-PAGE gelatin zymography as described previously (Bojić-Trbojević et al., 2019[8]). Zymograms were scanned and analyzed by the ImageMaster TotalLab v2.01 program (Amersham Biosciences Inc., USA).
HTR-8/SVneo tube formation
Tube formation assay was done as previously described with minor changes (Beltrame et al, 2018[7]). Briefly, 96-well plates were coated with 40 μl/well of growth factor-reduced Matrigel diluted with Opti-MEM to final concentration of 7.5 mg/ml and incubated 30 min at 37 °C to solidify. HTR-8/SVneo cells (4 x 104 cells/well) were seeded on coated wells in Opti-MEM 72 h after transfection. Images of tube structures were taken after 6 h of incubation using inverted light microscope (x10 magnification, Nikon TMS, Nikon Instruments, Inc., USA) and digital camera (Canon, PowerShot S50, Canon Inc., Japan). Four non-overlapping fields of view per well were analyzed. Total tube length and number of branching points were quantified using Image J software (National Institutes of Health, USA). The branching points were considered as a point from which two or more tubes branched.
HUVEC viability test and determination of adherent cell number
The viability and adherent cell number of HUVEC were assessed using the MTT test or crystal violet staining as previously described with minor changes (Stefanoska et al., 2013[33]). HUVEC were seeded in gelatin-coated 96-well plates (2 x 104 cells/well) in 100 μl of HUVEC medium and maintained overnight in a humidified chamber. Cells were rinsed and incubated for 24 h in a mixture of conditioned media of transfected HTR-8/SVneo cells collected 72 h after transfection and HUVEC medium, ratio 2:1, 100 μl/well. For MTT test, 10μl of MTT (5 mg/ml) was added to each well upon treatment. After incubation for 2 h at 37 ̊ C, medium was replaced by 1-propanol. Absorbance was measured at 540 nm using a microplate reader (LKB, Austria).
For crystal violet staining, the cells were dried, fixed and stained with 0.05 % crystal violet in 25 % methanol. The incorporated dye was dissolved in 0.1 M sodium citrate in 50 % ethanol. Optical density was read at 540 nm. The results of three experiments with six replicates are presented as a percentage of lipofectamine control values.
Trophoblast integration into endothelial cell monolayers
Trophoblast-endothelial cell monolayer co-culture was used to investigate ability of HTR-8/SVneo cells to integrate into endothelium and replace the endothelial cells, as previously described with minor modifications (Bainbridge et al., 2009[6]). Briefly, HUVEC cells were seeded onto gelatin coated 13 mm glass coverslips and grown in HUVEC medium to full confluence. HTR-8/SVneo cells were collected 72 h after transfection, labelled with 15 μM Cell Tracker Blue CMAC dye (Molecular Probes, Invitrogen, Thermo Fisher Scientific Inc., USA) according to the manufacturer's instruction and seeded onto HUVEC's monolayer at 7.5 x 104 cells/well/ 24-well plates in 1:1 mixture of HUVEC and HTR medium. After 24 h co-cultures were examined by Carl Zeiss Axio Imager.A1 microscope with AxioCam MRm camera (Carl Zeiss, Germany). Captures were taken with 10x objective all over the coverslips and HTR-8/SVneo cell integration into endothelial cell monolayers was quantified as a percentage of total field area occupied by trophoblast cell islands (blue labeled) using Image J software (National Institutes of Health, USA).
Statistical analyses
The data was analyzed using GraphPad Prism Demo Software (GraphPad Software, Inc., USA). One-way analysis of variance (ANOVA) with Tukey post-hoc test (α = 0.05) was used for statistical analyses since data passed normality test. Values were considered significantly different when p < 0.05. All experiments were done at least three times in duplicate and the mean values for the lipofectamine control was set to the 100 % and the data were presented as percentage of the lipofectamine control unless otherwise stated.
Results
MIF specific siRNA decreases MIF expression, secretion and Matrigel invasion by HTR-8/SVneo cells
Efficiency of MIF silencing was verified at mRNA (Figure 1a(Fig. 1)) and protein levels in whole cell lysates (Figure 1b, c(Fig. 1)) and in conditioned media (Figure 1c, d(Fig. 1)). MIF mRNA expression was reduced to 15 % of control after 48 h and to 3 % of control after 72 h of culture (Figure 1a(Fig. 1), p<0.001). In whole cell lysates MIF protein was reduced to 45 % and 38 %, at 48 h and 72 h after transfection, respectively (Figure 1b(Fig. 1); p<0.001). Secreted MIF, detected in cell conditioned media was reduced to 63 % (p<0.01) and 37 % (p<0.001) of lipofectamine control, after 48 h and 72 h respectively (Figure 1d(Fig. 1)). Silencing of MIF had no effect on HTR-8/SVneo cell viability neither 48 h nor 72 h following transfection (Figure 1e(Fig. 1)). The importance of endogenous MIF for trophoblast cell function was studied using in vitro Matrigel invasion assay. HTR-8/SVneo cells, 48 h following transfection, had reduced capacity for Matrigel invasion down to 59 % of control (Figure 1f(Fig. 1); p<0.01).
The effect of MIF silencing on the expression of integrins and MMPs in HTR-8/SVneo cells
Possible mediators of reduction in invasive capacity of HTR-8/SVneo cells were sought among integrin subunits and MMPs. Integrin α1 was reduced to 86 % of control (Figure 2a(Fig. 2); p<0.01), as shown by flow cytometry. Gelatin zymography revealed that both tested MMPs - MMP2 and MMP9 were decreased to 87 % (p<0.05) and 57 % (p<0.001) of control, respectively (Figure 2b, c, d(Fig. 2)).
MIF silencing impairs endovascular differentiation of HTR-8/SVneo cells and integration into endothelial cell monolayer
The importance of MIF for differentiation of HTR-8/SVneo cells to endovascular trophoblast-like phenotype was studied using tube formation assay on Matrigel (Figure 3a(Fig. 3)). HTR-8/SVneo cells display tube formation ability in culture (Highet et al., 2012[15]). Knockdown of MIF decreased cell ability to form tubes. The total tube length was decreased to 68.6 % (32397±1137 and 22218±1352 pixels for L and siMIF respectively, p<0.001) (Figure 3b(Fig. 3)), while the number of branching points was reduced to 57.8 % of lipofectamine control (99.6±3.8 and 57.6±3 branching points for L and siMIF respectively, p<0.001) (Figure 3c(Fig. 3)).
Interaction of trophoblast and endothelium was studied using direct co-culture of HTR-8/SVneo cells and HUVEC. MIF knockdown decreased the capacity of HTR-8/SVneo cells to replace HUVEC cells to 73 % of control (p<0.001, Figure 4a, b(Fig. 4)). The possibility that secreted HTR-8/SVneo cell factors modulate HUVEC survival was studied using conditioned media of transfected HTR-8/SVneo cells in MTT and adherent cell number assays of HUVEC. These media had no effect on HUVEC survival (Figure 4c, d(Fig. 4)).
The expression of N-cadherin, as possible mediator of reduction in HTR-8/SVneo cell integration into HUVEC monolayer, was studied by flow cytometry. The expression of this adhesion molecule was reduced to 63 % of control (p<0.05, Figure 4e(Fig. 4)) after knockdown of MIF.
Discussion
Implantation of the human embryo and placentation takes place in a proinflammatory environment. Many of the cytokines/chemokines contribute to these processes by stimulating or limiting trophoblast invasion (Lash, 2015[23]; Pollheimer et al., 2018[31]). MIF is abundantly present at the feto-maternal interface, produced by trophoblast itself, as well as various maternal cell types such as decidual stromal cells and immune cells (Arcuri et al., 2001[4]). Our previous results showed that blocking of MIF by ISO-1 decreased trophoblast cell migration and invasion, while addition on rhMIF had the opposite effect (Jovanović Krivokuća et al., 2015[19]). ISO-1 is a small chemical inhibitor designed to block tautomerase activity of MIF, previously shown essential for its proinflammatory action (Al-Abed et al., 2005[1]). Here, the role of endogenous trophoblast MIF was further investigated, and its relevance for endovascular trophoblast cell function in particular. The data presented here show that attenuation of endogenous MIF decreased HTR-8/SVneo cell invasion, which is in keeping with the previously reported reduction observed when extracellular MIF tautomerase activity was neutralizated with ISO-1. Taken together, MIF is shown to act both as an autocrine stimulator of trophoblast invasion, but also in a paracrine manner, since our previous results showed that addition of ISO-1 to decidual stromal cell conditioned media decreased their pro-invasive action on trophoblast (Jovanović Krivokuća et al., 2015[19]). Other cell types have also shown reduced migratory and/or invasive capacity when MIF activity was abolished after knockdown of MIF by specific siRNA, such as endometrial cancer cell line HEC-1A (Md Fuzi et al., 2018[26]), oral squamous cell carcinoma (Zeng et al., 2016[37]), mouse colorectal cancer cell line CT-26 (Wu et al., 2017[36]). Both siRNA-mediated silencing of MIF, and the use of specific inhibitors led to a significant decrease in the invasive ability of the gall bladder cancer cell lines (Subbannayya et al., 2015[34]). Inhibition of lung adenocarcinoma cell line invasion by ISO-1 as well as MIF siRNAs was also reported (Rendon et al., 2007[32]).
It has been well documented that as EVT acquires invasiveness, changes in integrin repertoire (loss of α6β4, and upregulation of α1β1and α5β1) occur (Damsky et al., 1992[11]). Moreover, invasive EVT produces extracellular matrix-degrading enzymes, such as MMPs (Lala and Graham, 1990[22]; Librach et al., 1991[24]). Previously, we have shown that blocking MIF by ISO-1 decreased integrin α1 (Jovanović Krivokuća et al., 2015[19]), which was replicated here with MIF siRNA approach. Attenuation of endogenous MIF had negative effect on gelatinolytic activity of MMP2 and MMP9. This reduction could contribute to reduced Matrigel invasion of MIF silenced HTR-8/SVneo cells, since this process requires action of proteolytic enzymes. Similarly, blocking MIF by ISO-1 in our previous study also involved reduction in MMP2 and MMP9 (Jovanović Krivokuća et al., 2015[19]). Not much is currently known regarding the influence of MIF inhibition by siRNA on integrin and MMP expression. Integrin β1 was supressed in mouse colon 26 cells (Sun et al., 2005[35]), while MMP9 was reduced by MIF siRNA in oral squamous carcinoma cells (Zeng et al., 2016[37]).
The main focus of our study was to establish whether trophoblast MIF might be involved in spiral artery remodeling process. The data presented here show that MIF may be an important factor in trophoblast differentiation to endovascular phenotype, since HTR-8/SVneo cells were significantly less capable of tube formation on Matrigel after knockdown of this cytokine. The mechanism of endothelium replacement by trophoblast is still not completely elucidated, but it is known to involve both endothelium apoptosis and regression (Ashton et al., 2005[5]; Pijnenborg et al., 2006[29]). Both invading trophoblast and vascular cells produce proteolytic enyzmes, enabling degradation of subendothelial basement membrane. Trophoblast and vascular smooth muscle cells were suggested to act cooperatively to degrade elastin in basal membrane of the vessel, by producing elastase MMP-12 (Harris et al., 2010[14]). In our study, HUVEC cells were used as model for endothelium. Previous study has reported that there was no difference between HUVEC and decidual endothelial cells with respect to response to trophoblast (Ashton et al., 2005[5]). Using co-culture of HTR-8/SVneo cells with HUVEC, we have shown that trophoblast MIF might be involved in remodeling process, since MIF silencing significantly decreased trophoblast capacity to integrate into HUVEC monolayers. Secreted factors dependent on trophoblast MIF, do not change HUVEC viability, as conditioned media of MIF knockdown HTR-8/SVneo cells had no effect. Changes in the expression of various adhesion molecules occur during interstitial and endovascular invasion (Damsky et al., 1992[11]; Damsky and Fisher, 1998[10]; Zhou et al., 1997[39]). Endovascular trophoblast was shown to express endothelial adhesion molecules such as VE-(endothelial) cadherin, platelet-endothelial adhesion molecule-1, vascular endothelial adhesion molecule-1, and a4-integrins, and integrin αVβ3 (Zhou al., 1997[39]). Recently, N-cadherin was reported essential for the interaction of trophoblast and endothelium (Multhaup et al., 2018[27]). This cadherin was specifically expressed in HTR-8/SVneo cell line, which showed much more pronounced interaction with pre-formed HUVEC networks compared to other used trophoblast cell lines, Jeg-3, AC-1M32 and AC-H3P (Multhaup et al., 2018[27]). Knockdown of N-cadherin by specific siRNA in HTR-8/SVneo cells led to disruption of interaction with endothelium networks (Multhaup et al., 2018[27]). We therefore wondered whether this protein might be mediating the reduction in HTR8/SVneo integration into HUVEC monolayer observed here. Indeed, this hypothesis was confirmed, since MIF specific siRNA treatment resulted in decreased expression of N-cadherin. However, potential action of other mediators is not excluded, and merrits further attention.
Taken together, our results confirm that MIF participates in multiple early pregnancy events necessary for normal implantation and placentation, and show that trophoblast MIF significantly contributes to various aspects of trophoblast function.
Acknowledgements
This work was funded through project 173004 of the Ministry of Education, Science, and Technological Development, Republic of Serbia.
Conflict of interest
The authors declare that they have no conflict of interest.
References
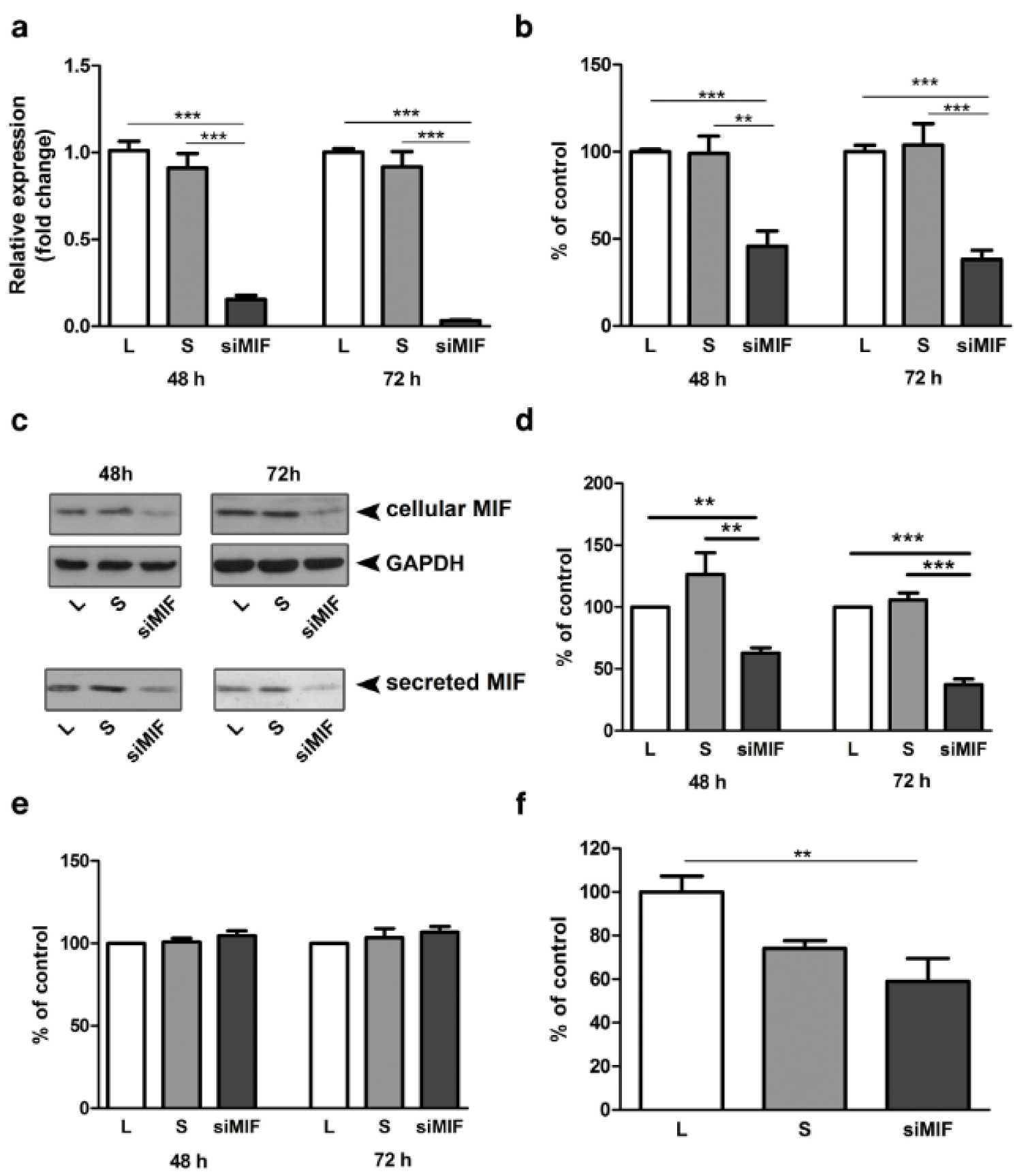
Figure 1: MIF specific siRNA reduces MIF expression and cell invasion of HTR-8/SVneo cells. MIF specific siRNA effectively reduced mRNA (a) and protein expression (b, c, d) in whole cell lysates (b, c) and secreted MIF in conditioned media (c, d). Representative Western blots are shown in c. Inhibition of MIF expression had no effect on cell viability (e), but led to a significant decrease in HTR-8/SVneo cell invasion in Matrigel invasion assay (f). Data are presented as mean +SEM, ** p<0.01, *** p<0.001. n = 5 (a), n = 3 (b, f), n=4 (d, e)
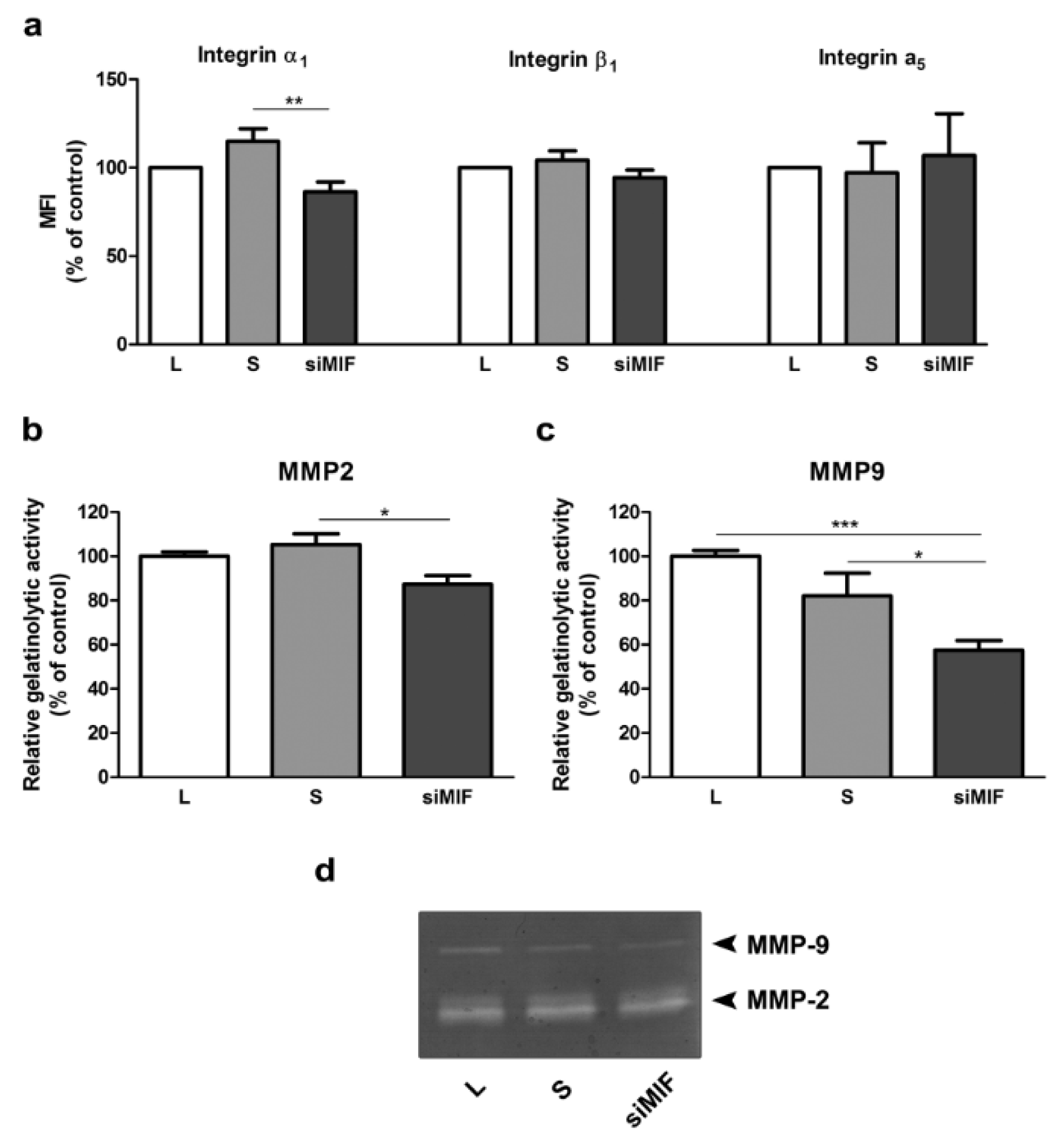
Figure 2: The effect of MIF knockdown by specific siRNA on mediators of trophoblast invasion in HTR-8/SVneo cells. Integrin subunit α1 was significantly reduced, while subunits α5 and β1 were not changed as assessed by flow cytomerty (a). Gelatine zymography showed a significant decrease in both MMP2 (b) and MMP9 (c) levels. Representative zymogram is shown in d. MFI - mean fluorescent intensity. Data are presented as mean +SEM, * p<0.05, ** p<0.01, *** p<0.001, n =3-6
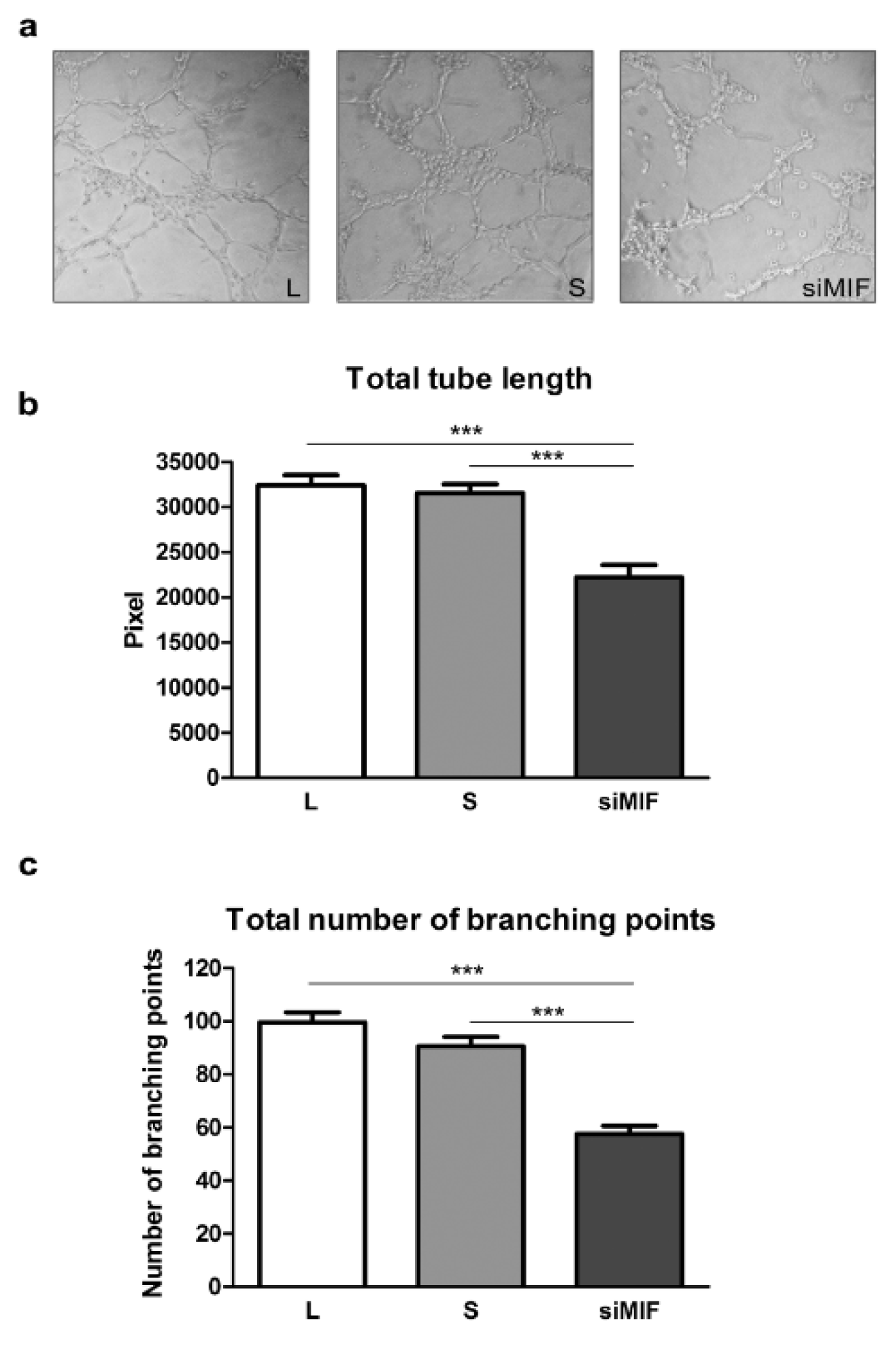
Figure 3: The effect of inhibition of MIF expression on HTR-8/SVneo cell acquisition of endothelial-like phenotype in tube formation assay on Matrigel (a). Total tube length (b) and total number of branching points (c) were significantly reduced. Data are presented as mean +SEM, *** p<0.001. n = 4
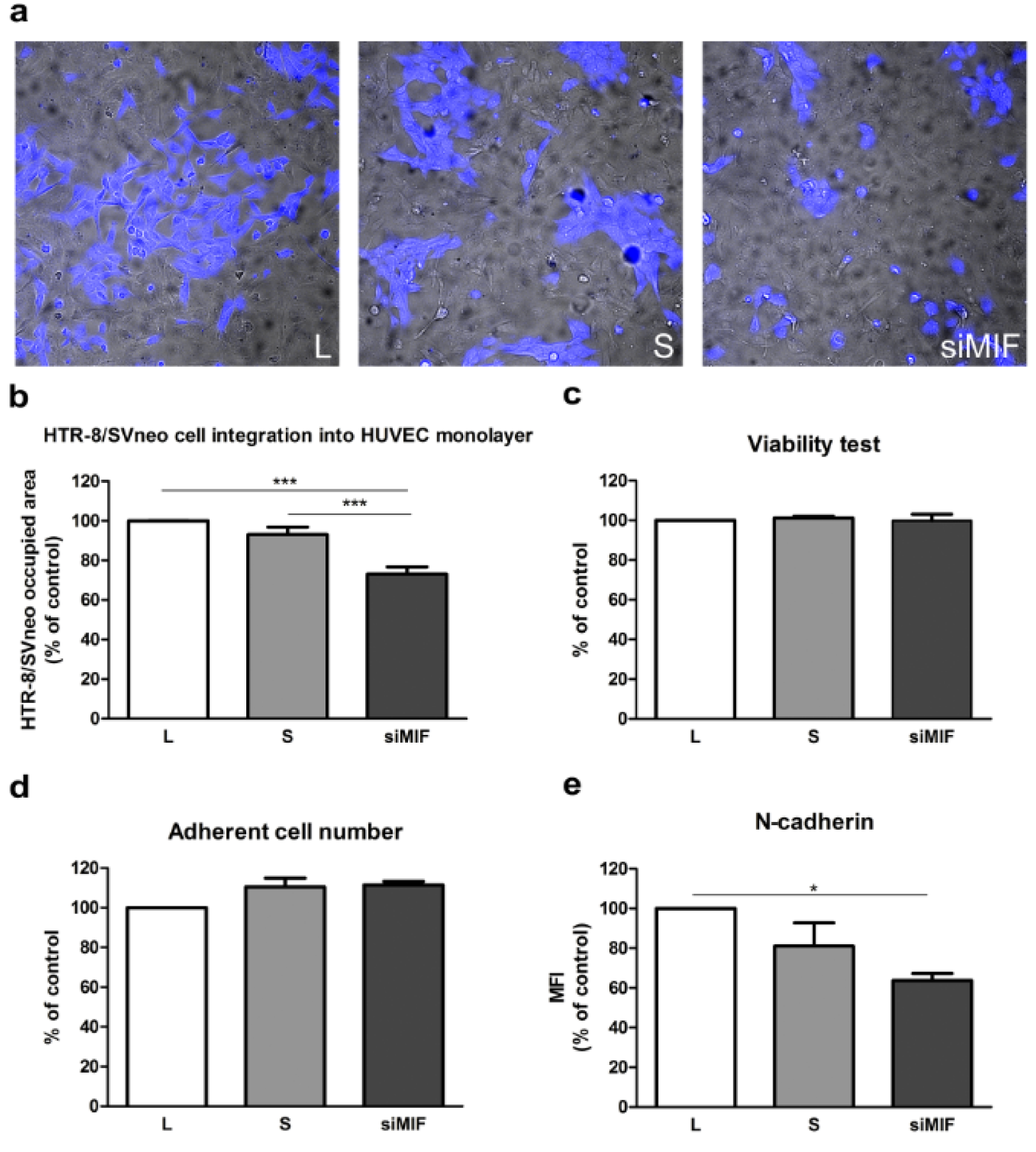
Figure 4: MIF specific siRNA decreases endothelium replacement capacity of HTR-8/SVneo cells. Inhibition of MIF expression by siRNA (72 h) led to a significant reduction in the capacity of HTR-8/SVneo cells to replace endothelium in trophoblast-HUVEC co-culture model (a, b). Conditioned media from MIF-silenced HTR-8/SVneo cells had no significant effect on HUVEC cell viability in MTT test (c), or adherent cell numbers (crystal violet staining) compared to control. N-cadherin expression was significantly reduced in HTR-8/SVneo cells following MIF knockdown as assessed by flow cytometry (e). MFI - mean fluorescent intensity. Data are presented as mean +SEM, * p<0.05, *** p<0.001. n = 4 (b), n = 3 (c, d)
[*] Corresponding Author:
Milica Jovanovic Krivokuca, Institute for the Application of Nuclear Energy, INEP, University of Belgrade, Banatska 31b, 11080 Belgrade, Serbia; Tel. +381 11 316 90 58, Fax. +381 11 2618 724, eMail: milicaj@inep.co.rs