Research article
Rational design of novel coumarins: A potential trend for antioxidants in cosmetics
Apilak Worachartcheewan1[*], Veda Prachayasittikul2, Supaluk Prachayasittikul2, Visanu Tantivit1, Chareef Yeeyahya1, Virapong Prachayasittikul3
1Department of Community Medical Technology, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand2Center of Data Mining and Biomedical Informatics, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand
3Department of Clinical Microbiology and Applied Technology, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand
EXCLI J 2020;19:Doc209
Abstract
Coumarins are well-known for their antioxidant effect and aromatic property, thus, they are one of ingredients commonly added in cosmetics and personal care products. Quantitative structure-activity relationships (QSAR) modeling is an in silico method widely used to facilitate rational design and structural optimization of novel drugs. Herein, QSAR modeling was used to elucidate key properties governing antioxidant activity of a series of the reported coumarin-based antioxidant agents (1-28). Several types of descriptors (calculated from 4 softwares i.e., Gaussian 09, Dragon, PaDEL and Mold2 softwares) were used to generate three multiple linear regression (MLR) models with preferable predictive performance (Q2LOO-CV = 0.813-0.908; RMSELOO-CV = 0.150-0.210; Q2Ext = 0.875-0.952; RMSEExt = 0.104-0.166). QSAR analysis indicated that number of secondary amines (nArNHR), polarizability (G2p), electronegativity (D467, D580, SpMin2_Bhe, and MATS8e), van der Waals volume (D491 and D461), and H-bond potential (SHBint4) are important properties governing antioxidant activity. The constructed models were also applied to guide in silico rational design of an additional set of 69 structurally modified coumarins with improved antioxidant activity. Finally, a set of 9 promising newly design compounds were highlighted for further development. Structure-activity analysis also revealed key features required for potent activity which would be useful for guiding the future rational design. In overview, our findings demonstrated that QSAR modeling could possibly be a facilitating tool to enhance successful development of bioactive compounds for health and cosmetic applications.
Keywords: coumarin, antioxidant activity, rational design, QSAR, MLR
Introduction
Free radicals (or oxidants) are highly reactive molecules containing an unpaired electron, which are generated as by-products of physiological processes and intracellular pathways (Valko et al., 2007[50]; Winyard et al., 2005[52]). These oxidants are well-known for their harmful potential and deleterious effects in cellular components (i.e., DNA, proteins and lipids). In normal condition, these radicals are scavenged/neutralized by antioxidant defense mechanism (i.e., endogenous antioxidant molecules and antioxidant enzymes) to prevent cellular oxidative damages. However, the shift of oxidative balance occurs in a condition whereby radicals are overproduced or antioxidant defense mechanism is depleted. This situation leads to excessive accumulation of free radicals and oxidative stress. Oxidative damage involves in pathogenesis and progression of many chronic and aging diseases (i.e., cancer, diabetes mellitus, neurodegenerative diseases, and cardiovascular diseases) (Valko et al., 2007[50]; Winyard et al., 2005[52]). Furthermore, free radicals have been recognized as one of the factors contributing to aging skin (Bogdan Allemann and Baumann, 2008[5]). Antioxidant compounds have been well-recognized for their wide-ranging health applications, especially in cosmeceutical area. Currently, an addition of antioxidants as active ingredients in cosmetics and personal care products has been widely documented (Kusumawati and Indrayanto, 2013[21]; Lupo, 2001[23]). Therefore, discovery of novel potent antioxidant compounds, both from chemical synthesis (Prachayasittikul et al., 2009[32]; Subramanyam et al., 2017[47]; Worachartcheewan et al., 2012[58]) and natural-derived sources (Elansary et al., 2018[9]; Krishnaiah et al., 2011[20]; Prachayasittikul et al., 2008[30], 2009[31], 2013[36]; Wongsawatkul et al., 2008[55]), has been noted to be an attractive research area, especially in cosmetic applications (Kusumawati and Indrayanto, 2013[21]; Lupo, 2001[23]).
Coumarins, known as benzopyrones, are natural secondary metabolites bearing fused benzene and α-pyrone rings (Witaicenis et al., 2014[53]). Natural-derived coumarins are found in a wide range of plants (Lee et al., 2007[22]; Rodríguez-Hernández et al., 2019[40]; Saleem et al., 2019[44]; Venditti et al., 2019[51]). Coumarins displayed a variety of biological activities including antimicrobial (Arshad et al., 2011[3]), antioxidant (Erzincan et al., 2015[10]), anticancer (Nasr et al., 2014[28]), and anti-inflammatory (Witaicenis et al., 2014[53]) activities. Although synthetic coumarins were banned for oral products due to their potential toxicities, they are attractive for topical uses due to their high skin penetrating property (Stiefel et al., 2017[46]). Additionally, coumarins are widely used as fragrance ingredient in cosmetics and personal care products because of their sweet herbaceous scent (Ma et al., 2015[24]; Stiefel et al., 2017[46]). Antioxidant property and protective effects against skin photo-aging of coumarins have also been remarked in cosmetic area (Kostova et al, 2011[19]; Lee et al., 2007[22]). Previously, a set of synthesized coumarin derivatives containing 2-methylbenzothiazolines, sulphonamides, and amides were reported to exhibit antioxidant activity with IC50 values range of 0.024-2.888 mM (Khoobi et al., 2011[18]; Saeedi et al., 2014[42]). However, deeper understanding of structure-activity relationships (SAR) and mechanism of action is still necessary for an effective rational design of coumarin-based antioxidant agents (Kostova et al., 2011[19]).
Computational approaches have been widely recognized to facilitate and increase success rate of drug development (Nantasenamat and Prachayasittikul, 2015[27]; Prachayasittikul et al., 2015[37]). Quantitative structure-activity relationship (QSAR) modeling is an in silico method to reveal the relationship between chemical structures of the compounds and their biological activities. QSAR modeling provides useful findings such as key features, properties, or moieties that are required for potent activity, which would benefit further rational design of the related compounds. Currently, success stories of QSAR-driven rational design of several classes of promising lead compounds have been documented for anticancer agents (Prachayasittikul et al., 2015[33]), aromatase inhibitors (Prachayasittikul et al., 2017[35]), and sirtuin-1 activators (Pratiwi et al., 2019[38]). In cosmetic area, QSAR modeling has been employed to improve understanding towards SAR of tyrosinase inhibitors (Gao, 2018[13]; Khan, 2012[17]).
Accordingly, this study aims to construct QSAR models to elucidate SAR of a set of antioxidant coumarin derivatives (1-28, Figure 1(Fig. 1)) originally reported by Khoobi et al. (2011[18]) and Saeedi et al. (2014[42]). Herein, QSAR models were constructed using multiple linear regression (MLR) algorithm to clearly demonstrate the linear relationship along with insight SAR analysis. In an attempt to find a robust and validating QSAR models, chemical descriptors were generated using different four softwares (i.e., Gaussian 09, Dragon, PaDEL and Mold2 softwares) to increase a variety of represented physicochemical properties. Consequently, an additional set of structurally modified compounds were rationally designed based on key findings of the constructed models, and their antioxidant activities were predicted to reveal the promising ones with potential for further synthesis and development.
Materials and Methods
Data set
A data set of twenty-eight coumarin-based antioxidants (1-28, Figure 1(Fig. 1)) was retrieved from the literature (Khoobi et al., 2011[18]; Saeedi et al., 2014[42]), in which their antioxidant activities are presented in Table 1(Tab. 1). All tested compounds were evaluated by 1,1-diphenyl-2-picryhydrazyl (DPPH) assay (detailed methodology is provided in original literatures (Khoobi et al., 2011[18]; Saeedi et al., 2014[42])). The activity was denoted as an IC50 value (mM) which indicates concentration of the compound which can inhibit 50 % of the generated DPPH radicals in experimental setting. As a part of data pre-processing, the unit of IC50 values was converted from mM to M, and the IC50 values were further transformed into pIC50 (−log IC50) by taking the negative logarithm to base 10 as shown in Table 1(Tab. 1). The compound with high pIC50 (low IC50) represented the high antioxidant activity. A schematic workflow of QSAR model development is provided in Figure 2(Fig. 2).
Molecular structure optimization
Molecular structures of the coumarin derivatives were constructed by GaussView (Dennington et al., 2003[7]), which were subjected to geometrical optimization by Gaussian 09 (Revision A.02) (Frisch et al., 2009[12]) at the semi-empirical level using Austin Model 1 (AM1) followed by density functional theory (DFT) calculation using Becke's three-parameter hybrid method and the Lee-Yang-Parr correlation functional (B3LYP) together with the 6-31 g(d) basis.
Descriptor calculation and feature selection
The physicochemical properties (i.e., quantum chemical and molecular descriptors) were generated by different calculating softwares including Gaussian 09, Dragon, version 5.5. (Talete, 2007[49]), PaDEL, version 2.20 (Yap, 2011[60]) and Mold2, version 2.0 (Hong et al., 2008[15]) softwares. The calculated descriptors as numerical values could be used to represent properties of the compounds, and were further used as predictors (X variables) for QSAR model construction. List of calculated descriptors are shown as follows.
Quantum chemical descriptors calculation obtained by low energy conformers from the geometrical optimization using Gaussian 09 composed of the total energy (Etotal) of the molecule, the highest occupied molecular orbital energy (EHOMO), the lowest unoccupied molecular orbital energy (ELUMO), the total dipole moment (μ) of the molecule, the electron affinity (EA), the ionization potential (IP), the energy difference of HOMO and LUMO (HOMO-LUMOGap), Mulliken electronegativity (χ), hardness (η), softness (S), electrophilicity (ω), electrophilic index (ωi), and the mean absolute atomic charge (Qm) (Worachartcheewan et al., 2014[57]). Furthermore, the output files from geometrical optimization of Gaussian 09 were used as the input data for calculating a set of 3,224 molecular descriptors using Dragon software. The calculated descriptors included 22 classes comprising 48 constitutional descriptors, 119 topological descriptors, 47 walk and path counts, 33 connectivity indices, 47 information indices, 96 2D autocorrelation, 107 edge adjacency indices, 64 burden eigenvalues, 21 topological charge indices, 44 eigenvalue based indices, 41 randic molecular profiles, 74 geometrical descriptors, 150 RDF descriptors, 160 3D-MoRSE descriptors, 99 WHIM descriptors, 197 GETAWAY descriptors, 154 functional group counts, 120 atom centered fragments, 14 charge descriptors, 29 molecular properties, 780 2D binary fingerprints, and 780 2D frequency fingerprints.
An additional set of molecular descriptors was calculated by PaDEL software to give 1,444 1D and 2D descriptors, and Mold2 software to generate 777 descriptors by encoding the 2D chemical structure information. Before the calculation, the molecular structures were saved to *.smi and then converted to *.mol files using OpenBabel version 2.3.2 (The Open Babel Package 2015). The *.mol files were used as the input data for calculation by PaDEL and Mold2 softwares.
Descriptors selection was performed to filter a set of important informative descriptors from a whole set of descriptors. Feature selection was initially performed by stepwise multiple linear regression (MLR) using SPSS statistics 18.0 (SPSS Inc., USA) followed by determination of intercorrelation using Pearson's correlation coefficient using cutoff value of |r| ≥ 0.9. Any pairs of descriptors with |r| ≥ 0.9 were defined as highly correlated predictors, and one of them was excluded.
Data splitting
The data set of coumarin derivatives (1-28) was randomly selected, in which 85 % (~23 compounds) of the original data set was used as the training and the leave one-out cross-validation (LOO-CV) sets, and 15 % (~5 compounds) was used as the external set. The training set was employed to generate the QSAR models, whereas LOO-CV and external sets were used to evaluate the models. LOO-CV method was performed for internal validation by excluding one sample out from the whole data set to be used as the testing set while the remaining N−1 samples were used as the training set (Prachayasittikul et al., 2014[34]). This sampling process was repeated iteratively until every sample in the data set was used as the testing set. The external sets were used to validate the models.
Multivariate analysis
QSAR models were generated using the MLR according to the equation 1.

where Y is the antioxidant activity (pIC50), B0 is the intercept, and Bn are the regression coefficients of the descriptors Xn. The MLR method was performed using Waikato Environment for Knowledge Analysis (Weka), version 3.4.5 (Witten et al., 2011[54]).
Models evaluation
Statistical parameters were used to evaluate predictive performance of the constructed QSAR models. The calculated parameters included correlation coefficient (R2), root mean squared error (RMSE), predictivity (Q2), variance ratio (F ratio), adjusted correlation coefficient (R2Adj), standard deviation (s) and predicted residual sum of squares (PRESS) (Saghaie et al., 2013[43]; Worachartcheewan et al., 2013[56]).
Results and Discussion
Molecular descriptors selection
Chemical structures of the compounds and their antioxidant activities (Table 1(Tab. 1)) were used for construction of predictive models. The compounds were geometrically optimized with semi-empirical method AM1 followed by DFT/B3LYP/6-31 g(d) basis using Gaussian 09 to obtain lower-energy conformers. The optimized compounds were extracted to obtain 13 quantum chemical descriptors. These compounds were subsequently used as input files for calculating an additional set of 3,224 molecular descriptors (0D-3D) using Dragon software. The calculated descriptors with constant values and multi-collinearity were determined and removed to give a final set of 1,489 descriptors. In addition, original molecular structures of compounds were saved as *.smi file format and were converted into *.mol files using OpenBabel version 2.3.2. These *.mol files then were used as input files for descriptors calculation using Mold2 and PaDEL softwares to obtain sets of 777 Mold2 2D descriptors and 1,444 PaDEL 0D-2D descriptors, respectively. Consequently, feature selection was performed to select a set of informative descriptors for the whole calculated set. Descriptors showing significant correlation with their bioactivities were selected using stepwise MLR. A set of 14 informative descriptors included 4 Dragon descriptors (i.e., nArNHR, ISH, B04[O-O] and G2p), 6 Mold2 descriptors (i.e., D467, D278, D491, D384, D580 and D461), 4 PaDEL descriptors (i.e., SHBint4, SpMin2_Bhe, MATS8e and SssCH2) were obtained. Definition and numerical values of important descriptors are shown in Tables 2(Tab. 2) and 3(Tab. 3), respectively. Furthermore, the intercorrelation matrix between pair of molecular descriptors was performed using Pearson's correlation coefficient (r) (Supplementary Tables 1-3Worachartcheewan_26022020_supplementary_information.pdf). Cutoff value of |r| ≥ 0.9 was used to determine the intercorrelation. The results showed that there was no intercorrelation within a set of selected descriptors as displayed by low |r| values ≤ 0.9, which suggested that each descriptor was independent from other descriptors. Finally, a set of 14 selected descriptors was further employed to construct 3 QSAR models (according to types of software used to calculate descriptor values) for predicting antioxidant activity of the coumarin derivatives.
QSAR models
Descriptors obtained from these softwares have been demonstrated for their successful QSAR modeling such as antioxidant (Alisi et al., 2018[1]; Rastija et al., 2018[39]), antimicrobial (Alyar et al., 2009[2]; Basic et al., 2014[4]; Podunavac-Kuzmanović et al., 2009[29]), anticancer (Sławiński et al., 2017[45]; Suvannang et al., 2018[48]) and antiviral (Duchowicz et al., 2018[8]; Saavedra et al., 2018[41]; Worachartcheewan et al., 2019[59]) activities. Herein, three models were separately constructed based on the types of key descriptors (i.e., model 1 Dragon descriptors, model 2 Mold2 descriptors, and model 3 PaDEL descriptors). A set of 14 selected informative descriptors (as independent variables, Table 2(Tab. 2)) and antioxidant activities (pIC50 values as dependent variables) of the studied compounds were included in the data sets for construction of QSAR models using Eq. (1). Before building the models, the data set of coumarin derivatives (1-28) was split into training, LOO-CV, and external sets. The training set was used to construct the model using MLR algorithm whereas both LOO-CV and external sets were utilized for validating the constructed models. Compounds 1, 6, 15, 21 and 27 were randomly selected to be used as external sets, while the remaining 23 compounds in the data sets (i.e., 2-5, 7-14, 16-20, 22-26 and 28) were employed as training set. As a result, three QSAR models (models 1-3) were successfully constructed for predicting antioxidant activities (pIC50 values) of the studied coumarin analogs.
Model 1 (Dragon descriptors)
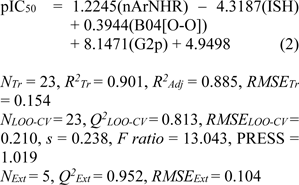
where NTr, NLOO-CV and NExt are the number of compounds of training, LOO-CV and external sets. R2Adj is the adjusted R2.
Four molecular descriptors calculated from Dragon software were used as predictors to construct QSAR model 1 as shown in Eq. (2). Statistical parameters indicating predictive performance of the model are summarized in Table 4(Tab. 4). Training set showed R2Tr = 0.901, RMSETr = 0.154, while the LOO-CV set displayed the Q2LOO-CV = 0.813, RMSELOO-CV = 0.210, and the external set with Q2Ext = 0.952, RMSEExt = 0.104. Comparative plot of experimental versus predicted antioxidant activities (pIC50) is shown in Figure 3a(Fig. 3). Residual values were calculated as a difference between experimental and predicted pIC50 values. Small residual values indicated the precision of model prediction. Plot of the residual values is shown in Figure 3b(Fig. 3).
Model 2 (Mold2 descriptors)
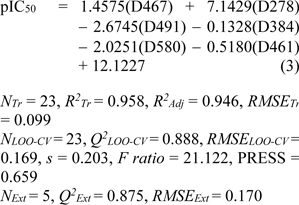
Six important descriptors obtained from the Mold2 software were used to generate the model 2 as shown in Eq. (3). Statistical analysis of the model is given in Table 4(Tab. 4). It was observed that the model provided statistical R2Tr = 0.958, RMSETr = 0.099 for the training set, Q2LOO-CV = 0.888, RMSELOO-CV = 0.169 for the LOO-CV set, and Q2Ext = 0.875, RMSEExt = 0.170 for the external set. The experimental versus the predicted antioxidant activities of the model 2 were graphically plotted (Figure 3c(Fig. 3)), while Figure 3d(Fig. 3) was the plot of residual values.
Model 3 (PaDEL descriptors)

Model 3 of Eq. (4) was built using 4 significant descriptors generated by PaDEL software. Statistical evaluation of the model is shown in Table 4(Tab. 4). The results showed that the QSAR model displayed R2Tr = 0.950, RMSETr = 0.109 for the training set, Q2LOO-CV = 0.908, RMSELOO-CV = 0.150 for the LOO-CV set, and Q2Ext = 0.885, RMSEExt = 0.166 for the external set. Comparison of experimental and the predicted antioxidant activities of model 3 are outlined in Figure 3e(Fig. 3), while residual values were displayed in Figure 3f(Fig. 3).
In overview, three constructed models provided satisfactory results as indicated by their statistical parameters such as R2, Q2, RMSE, F ratio and PRESS values. The R2 and Q2 of the obtained QSAR models were considered as acceptable values when R2>0.6 and Q2>0.5 (Golbraikh and Tropsha, 2002[14]; Nantasenamat et al., 2010[26]). These parameters of all constructed models were in acceptable range (model 1: R2Tr = 0.901, Q2LOO-CV = 0.813 and Q2Ext = 0.952, model 2: R2Tr = 0.958, Q2LOO-CV = 0.888 and Q2Ext = 0.875, model 3: R2Tr = 0.945, Q2LOO-CV = 0.908 and Q2Ext = 0.885. In addition, low values of RMSE, s and PRESS, but high value of F ratio indicated that the models were significant (RMSETr = 0.154, RMSELOO-CV = 0.210, RMSEExt = 0.104, s = 0.238, PRESS = 1.019 and F ratio = 13.043 in model 1, RMSETr = 0.099, RMSELOO-CV = 0.169, RMSEExt = 0.170, s = 0.203, PRESS = 0.659 and F ratio = 21.122 in model 2, and RMSETr = 0.109, RMSELOO-CV = 0.151, RMSEExt = 0.164, s = 0.170, PRESS = 0.518 and F ratio = 44.52 and in model 3) (Frimayanti et al., 2011[11]; Rastija et al., 2018[39]). The statistical (Table 4(Tab. 4)) and graphical (Figure 3(Fig. 3)) results showed that the QSAR models (models 1-3) gave a reliable agreement of the experimental and the predicted antioxidant values. Furthermore, the plots of experimental activity and residual values (Figures 3b, 3d and 3f(Fig. 3)) displayed the distribution of residuals on both sides of the zero values indicating that there are no systemic error in the models (Jalali-Heravi and Kyani, 2004[16]). Therefore, the QSAR models 1-3 could be possibly used and reliable for predicting the antioxidant activity of coumarin derivatives. Considering the correlation coefficient (Q2) of external set, it was shown that the Dragon descriptors gave the highest quality of the prediction for external test set (model 1: Q2Ext = 0.952) followed by the PaDEL descriptors (model 3: Q2Ext = 0.885) and the Mold2 descriptors (model 2: Q2Ext = 0.875).
Structure-activity relationship (SAR)
Regression coefficient values of the key descriptors (as independent variables) in QSAR models define the degree or weight of their influence on dependent variables (antioxidant activity: pIC50 values). According to the linear QSAR equations, high values of descriptors with positive regression coefficient and low values of descriptors with negative regression coefficient are required for potent antioxidant activity (high pIC50 values).
The descriptors in model 1 including nArNHR, B04[O-O] and G2p displayed positive values of regression coefficient involved in the increased antioxidant activity as a positive effect, while ISH descriptor having negative value of regression coefficient involved in the decreased activity as a negative effect. The important descriptors are ranked according to their regression coefficient values as G2p>nArNHR>B04[O-O]>ISH with corresponding values of 8.1471, 1.2245, 0.3944, and -4.3187, respectively. The descriptors in model 2 including D467 and D278 with positive regression coefficient values displayed the positive effect, whereas D491, D384, D580 and D461 descriptors exerted the negative effect on the bioactivity. The order of descriptors are D278>D467>D384>D461> D580>D491 with corresponding values of 7.1429, 1.4575, -0.1328, -0.5180, -2.0251, and -2.6745, respectively. For model 3, SHBint4, SpMin2_Bhe and SssCH2 descriptors showed the positive effect on activity, but MATS8e descriptor displayed the negative effect. The order of important descriptors are SpMin2_Bhe>SssCH2> SHBint >MATS8e with values of 6.9792, 0.3238, 0.2638 and -0.9768, respectively.
To gain insights into SAR, coumarin derivatives (1-28, Figure 1(Fig. 1)) are categorized into 3 groups according to their core structures (i.e., thiazole group I (1-9 and 18), sulfonamides group II (10-17) and amides group III (19-28) for effective SAR analysis. Thiazoles group I (1-9 and 18) showed antioxidant activity (Table 1(Tab. 1)) with pIC50 range of 2.539-4.612. The most potent and the least potent compounds of benzothiazoles group I were 5 (pIC50 = 4.612) and 6 (pIC50 = 2.539), respectively. Among group II compounds (10-17), compound 11 was the most active (pIC50 = 3.180), and 14 was the least active compound. For group III of amides 19-28, compound 21 displayed the most potent activity (pIC50 = 3.027) and compound 19 exhibited the lowest activity with pIC50 of 2.640.
It should be noted that coumarins substituted by thiazole at 3-position displayed better activity when compared with those substituted by sulfonamides and amides at 7- or 4- or 6- position (group II and III). The antioxidant activity (Table 1(Tab. 1)) is shown as the following trend: group I 5>3>4>1>2>18>8>7>9>6; group II 11>16>13>17>15>12>10>14; group III 21>28>22>20>23>26>27>24>25>19.
According to the significant descriptors in models 1-3, secondary (sec-) amine, polarizability, electronegativity and H-bond displayed positive effect in the antioxidant activity. This is noted in the most potent coumarin 5 bearing sec-amine (part of aromatic thiazole), and 7-OH group (on the coumarin ring) with H-bond and polarizability properties. On the other hand, tertiary (tert-) amine 6 without 7-OH group exerted the lowest activity among the coumarin derivatives 1-28. This could be implied that the sec-amine (-NH-) and OH as H-bond and polarizing group are important for the better activity.
In a series of compounds 1-28, only the sec-amines (1-5) had nArNHR = 1. The most potent compound 5 had higher values of nArNHR = 1, SHBint4 = 7.5875, G2p = 0.193, D467 = 0.805, but lower values of van der Waals volume (D491 = 0.471 and D461 = 0.186) when compared with the least potent compound 6 (nArNHR = 0, SHBint4 = 0.000, G2p = 0.169, D467 = 0.646, van der Waals volume: D491 = 0.805 and D461 = 0.520). Apparently, the tert-amines (6-8) and other amides, sulfonamides (10-28) as well as non-aromatic sec-amine 9 had nArNHR = 0.
Because each descriptor represents certain characteristic/property of the compound, QSAR equations are useful to guide effective rational molecular design of novel bioactive compounds with preferable activity (De et al., 2017[6]; Mitra et al., 2011[25]; Prachayasittikul et al., 2017[35]; Pratiwi et al., 2019[38]). To improve the antioxidant activity of these derivatives (1-28), 8 compounds (i.e., 2, 3, 4, 5, 11, 13, 18 and 21) were selected as parent compounds for structural modification. A series of novel analogs were rationally designed based on key influencing properties (which were revealed by descriptors presented in models 1-3). The modification was conducted by substitution of diverse types of functional groups with electronegativity, polarizability and H-bond properties (i.e., electron donating and electron withdrawing groups such as OH, NH2, SH, OCH3, CN, CF3 and halogen) on various positions of the coumarin ring. The new structurally modified compounds are shown in Supplementary Table 4Worachartcheewan_26022020_supplementary_information.pdf). Values of molecular descriptors of these compounds are provided in Supplementary Tables 5-7Worachartcheewan_26022020_supplementary_information.pdf). The prediction showed that the most potent compounds (pIC50) of each modified series were 2b, 2e (4.503, 4.502), 3n (6.340), 4g (6.445), 5h (7.016), 11a (4.526), 13d (4.356), 18c (4.197) and 21d (5.634) as shown in Figure 4(Fig. 4). All highly potent modified compounds were ranked as 5h>4g>3n>21d>11a>2b= 2e>13d>18c. The top three modified compounds (ranked as 5h>4g>3n) shared a common feature of 4-amino coumarin moiety (Figure 4(Fig. 4)). This moiety involves in H-bonding, polarizability, and electronegativity properties of the compounds which are essential for potent predicted activity. Apparently, these most potent compounds displayed the highest SHBint4 values of 16.903, 13.660, and 13.064 for 5h, 4g, and 3n, respectively.
The following discussed compounds are shown in Supplementary Table 4Worachartcheewan_26022020_supplementary_information.pdf. Compound 2 (pIC50 = 3.332) was modified by substitution of R1 (6-position) and R2 (5-position) groups (H, F, Cl, CF3, CN, NO2) on the core structure to give a new series of compounds 2a-2h with improved activity (pIC50 = 3.167-4.503), except for compounds 2f and 2h. Compounds 2b (5-CF3, 6-H) and 2e (5-CF3, 6-NO2) were the most potent compounds with comparable pIC50 values of 4.503 and 4.502, respectively. These could be due to the enhancing effect of 5-CF3 (R2) and 6-NO2 (R1) groups that provided high polarizability (G2p: 2b = 0.1820, 2e = 0.1730), electronegativity (SpMin2_Bhe: 2b = 1.847, 2e = 1.847), and potential H-bond (SHBint4: 2b = 5.150, 2e =5.480), thus, improved antioxidant activity of the modified compounds comparing with their parent 2 (G2p = 0.164, SpMin2_Bhe = 1.8175, SHBint4 = 4.0816).
Similarly, modified compounds 3a-3p were obtained by substitution of R1, R2, R3 at positions 7, 6, and 4 of the parent compound 3 (R1 = H, OH, OCH3, NH2, N(CH3)2, SH, SC6H5; R2 = F, Cl, Br, CF3; R3 = H, OH, OCH3, NH2, N(CH3)2). Compound 3n (R1 = H, R2 = Br, R3 = NH2) was predicted as the most potent one with pIC50 value of 6.340. Compounds 3f-3p (pIC50 = 4.129-6.340) were mostly active than the parent compound 3 (pIC50 = 4.003). It should be noted that these compounds (3f-3p) contained the same type of substituted R2 (Br) group as their parent 3 (6-Br). Therefore, higher H-bonding potential (SHBint4 = 13.064) and electronegativity (D467 = 0.736) play an important role in improving the activity of compound 3n when compared with its parent compound (3: SHBint4 = 4.2075, D467 = 0.722). The higher SHBint4 and D467 of 3n may result from the property of the substituted NH2 group in forming H-bond with the interacting species. In addition, the substitution with high electronegativity Br group (R2) at position 6 is required for improving activity of the compounds as noted for compound 3f-3p and the parent compound 3.
Structural modification improved activity of most of the compounds in series 4 (4a-4i: pIC50 = 4.033-6.445), except for compounds 4d and 4h.The results showed that 4g (R1 = 7-H, R2 = 4-NH2) was the most potent compound (pIC50 = 6.445). Comparing with the parent compound 4 (pIC50 = 3.973, SHBint4 = 4.442, G2p = 0.174), the most potent derivative 4g displayed higher potential H-bond (SHBint4 = 13.660) and higher polarizability (G2p = 1.960). It is reasonable to explain that 4-NH2 (R2) of 4g participates in H-bond forming and polarization through a keto group of the coumarin ring as shown by the resonant ionic form (4A) in Figure 5(Fig. 5). On the other hand, compound 4c (R1 = 7-NH2, R2 = 4-H) exerted lower activity with lower descriptors values (pIC50 = 0.4772, SHBint4 = 7.432, G2p = 0.1830) comparing with the most potent compound 4g.
The most potent coumarin 5 was modified to give derivatives 5a-5l, in which compounds 5f-5i displayed the improved antioxidant activity (pIC50 = 4.745-7.016). Compound 5h was the most potent one (pIC50 = 7.016) with higher H-bond potential (SHBint4 = 16.903), but lower electronegativity (MATS8e = 0.063) when compared with its parent 5 (pIC50 = 4.612, SHBint4 = 7.5875, MATS8e = 0.0848).
The amides 11, 13, and 21 were structurally modified by removing oxyketo group from the parent core structures to give sec-amines. The amine derivatives 11a-11f with improved activity (pIC50 = 3.402-4.526) were achieved from compound 11 (pIC50 = 3.180). When compared with its parent (amide 11: nArNHR = 0, D467 = 0.479, SHBint4 = 0.000, D491 = 0.688, D461 = 0.544 and MATS8e = 0.1872), improved activity of the most potent amine 11a (pIC50 = 4.526) was governed by its higher nArNHR = 1, electronegativity (D467 = 0.690), and H-bond (SHBint4 = 2.048), but lower van der Waals volume (D491 = 0.342 and D461 = 0.022) and electronegativity (MATS8e = -0.058). This could suggest that the improved activity of compounds requires a smaller size sec-amine (11a) with lower van der Waals volume but higher potential H-bonding. Similarly, a series of sec-amines 13a-13d (R1 = CH3, OH, OCH3, R2 = CH3, OCH3) were obtained (pIC50 = 3.341-4.356) from the amide 13 (pIC50 = 3.022). Compound 13d (pIC50 = 4.356) was the most potent amine with higher nArNHR = 1, D467 = 0.640, SHBint4 = 4.368, but lower van der Waals volume D461 = 0.455 and MATS8e = -0.125 comparing to its parent compound 13 (nArNHR = 0, D467 = 0.503, SHBint4 = 0.000, D461 = 0.515 and MATS8e = 0.1400). The amide 21 (pIC50 = 3.027) was modified to give analogs with improved antioxidant effect i.e., sec-amines analogs 21a-21h (pIC50 = 3.549-5.634). The most potent modified amine 21d (pIC50 = 5.634) displayed higher nArNHR = 1, electronegativity D467 = 0.657, H-bond SHBint4 = 9.823, electronegativity SpMn2-Bhe = 1.843 and SssCH2 = 0, but lower ISH = 0.869, van der Waals volume D491 = 0.350 and D461 = 0.436, and MATS8e = -0.219 when compared with the parent 21 (nArNHR = 0, D467 = 0.489, SHBint4 = 0.000, SpMin2-Bhe = 1.8395, SssCH2 = -0.1139, ISH = 0.923, van der Waals volume D491 = 0.82 and D461 = 0.612, and MATS8e = -0.0511).
Benzothiazole coumarin 18 was structurally modified to provide compounds 18a-18f (R1 = OH, OCH3, NH2, N(CH3)2, SH, SC6H5 at position 7). Comparing with the parent compound 18 (pIC50 = 3.260), compounds 18b-18e were more potent antioxidants (pIC50 = 3.758-4.197). Improved activity of the most potent compound 18c (pIC50 = 4.197) was governed by higher electronegativity (D467 = 0.595), total information content (D278 = 0.473) and H-bond (SHBint4 = 2.894), but lower van der Waals volume (D491 = 0.377and D461 = 0.299) when compared to parent compound 18 (D467 = 0.501, D278 = 0.413, SHBint4 = 0.000, D491 = 0.472 and D461 = 0.471).
It should be noted that the most potent modified compounds had higher values of H-bonding descriptor (SHBint4 = 2.048-16.903, Supplementary Table 7Worachartcheewan_26022020_supplementary_information.pdf) when compared with their parent compounds (SHBint4 = 0.000-7.5875, Table 3(Tab. 3)). Thus, SHBint4 might be the important descriptor in governing the potent antioxidant activity.
Conclusion
Understanding SAR is important for improving bioactivities and pharmacokinetic properties in development of potent and safe cosmetic products. Herein, a set of coumarin derivatives (1-28) with antioxidant activity was used to construct three QSAR models (1-3) using three different descriptor types and MLR method. Results of statistical evaluation showed that three generated QSAR models provide good reliability and comparable predictive performance (Q2LOO-CV = 0.813-0.908; RMSELOO-CV = 0.150-0.210; Q2Ext = 0.875-0.952; RMSE Ext = 0.104-0.166). In addition, good correlation obtained from model prediction suggests that the selected significant descriptors were shown to be good representatives for revealing correlation between chemical structures of the compounds (i.e., nArNHR, H-bonding, polarizability, van der Waals volume and electronegativity properties) and their antioxidant activities. An application of the constructed models was demonstrated by rationally designed an additional set of 69 structurally modified coumarins based on key descriptors, in which their antioxidant activities were predicted using the obtained QSAR models (1-3). Most of the rationally designed compounds displayed more improved antioxidant activity when compared with their parents. Particularly, the top three newly designed compounds (5h, 4g and 3n) showing high H-bonding (SHBint4) descriptor values which may play part in governing the most improved antioxidant activity. Finally, a set of newly designed promising coumarin analogs were highlighted for their potential to be further developed as potent antioxidants. Insights SAR findings also provided beneficial guidelines for the rational design of novel coumarin-based compounds with potent antioxidant effect for cosmetic applications.
Supplementary information
Supplementary informationWorachartcheewan_26022020_supplementary_information.pdf is available on the EXCLI Journal website.
Notes
Apilak Worachartcheewan and Supaluk Prachayasittikul (Center of Data Mining and Biomedical Informatics, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand; Phone: (662) 441-4376, Fax: (662) 441-4380, supaluk@g.swu.ac.th) contributed equally as corresponding authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This work was supported by the Thailand Research Fund (Grant No. MRG6180053) and the annual budget grant from Mahidol University (B.E. 2562-2563).
References
File-Attachments
- Worachartcheewan_26022020_supplementary_information.pdf (69,71 KB)
Supplementary information
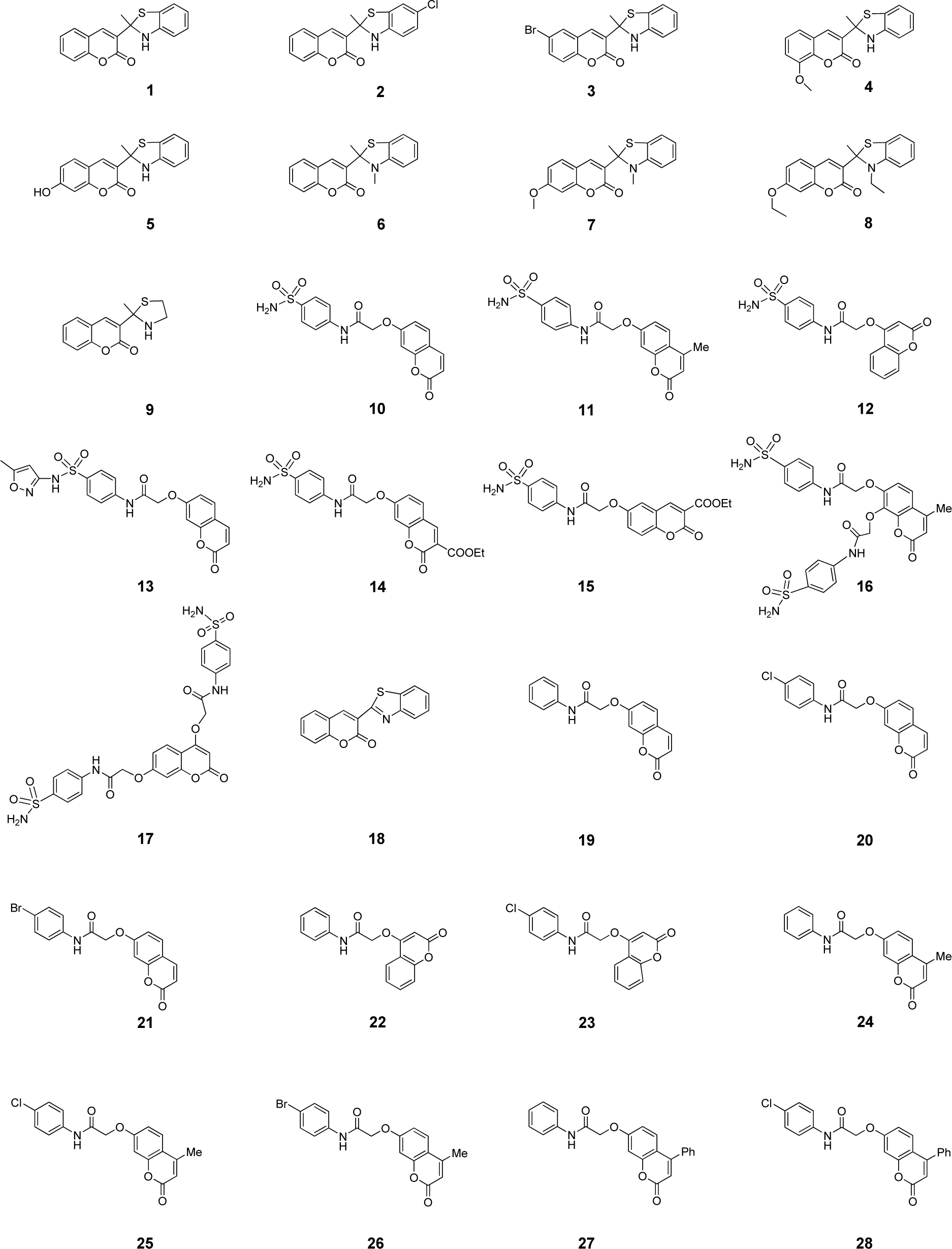
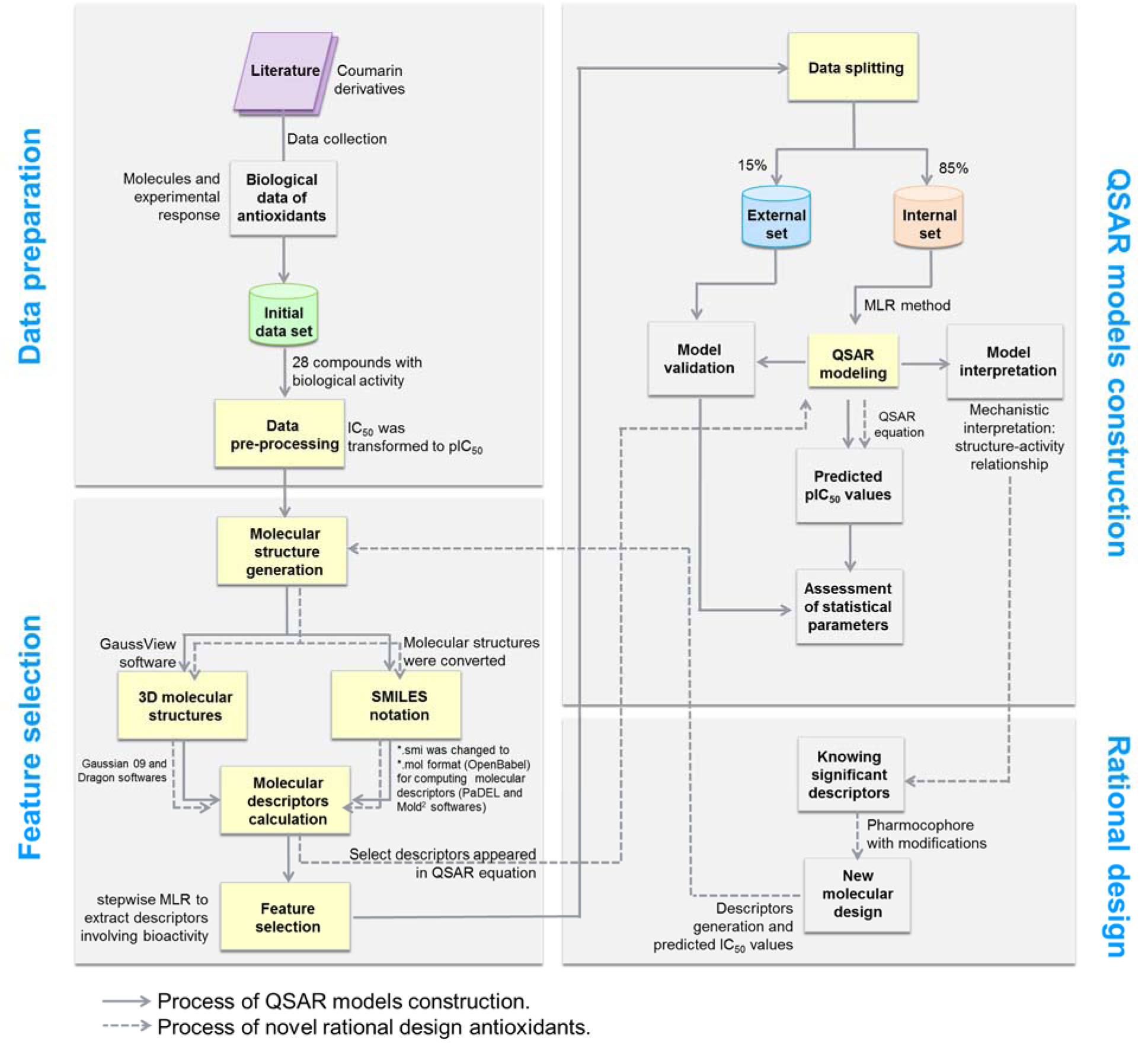
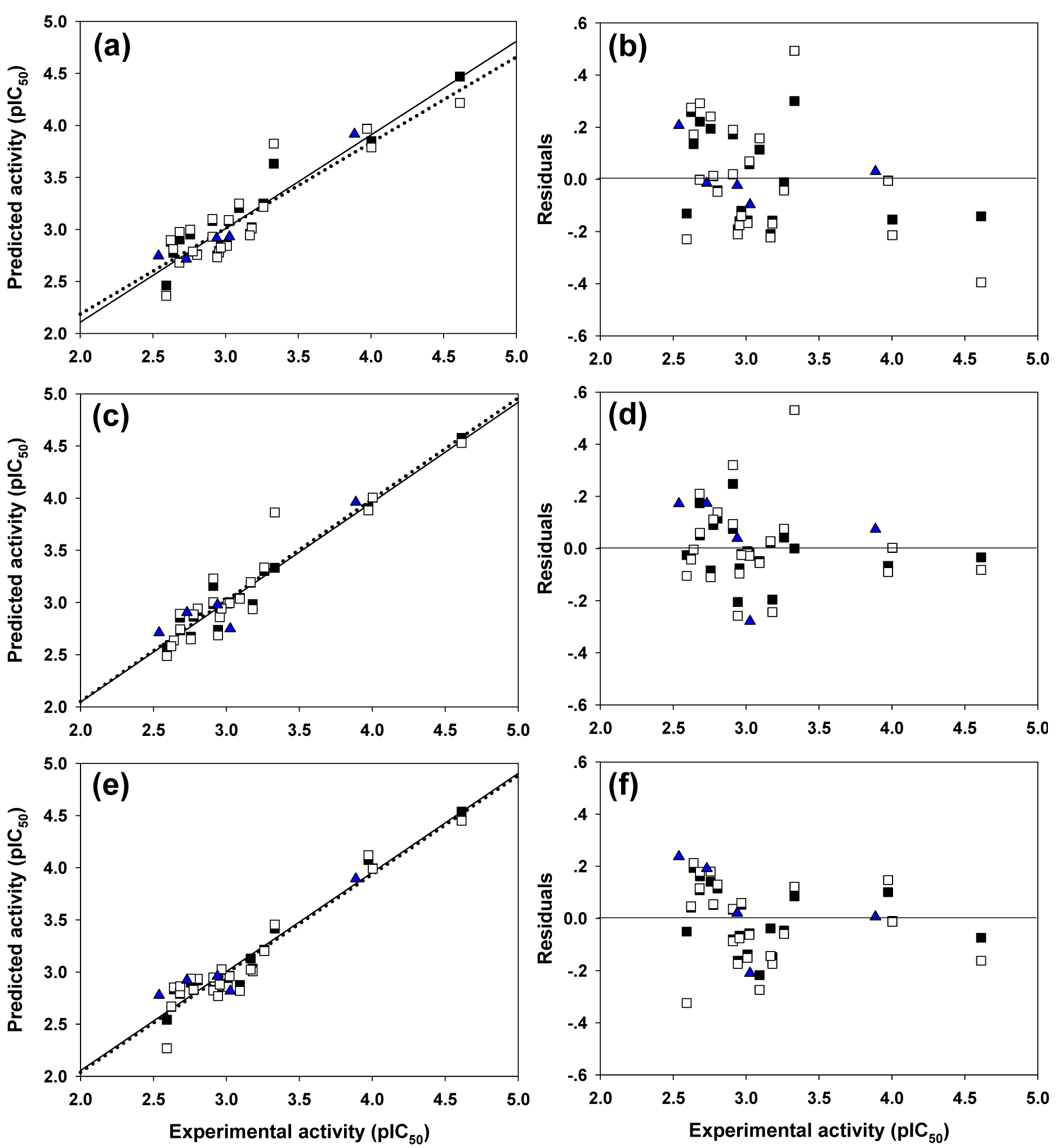
Figure 3: Plots of the experimental and the predicted activities of model 1 (3a), model 2 (3c), and model 3 (3e) for the training set (□; regression line is represented as solid line), the leave-one out cross-validated (■; regression line is represented as dotted line), and the external (▲) sets. Distribution of the experimental activity and residual values (the difference between experimental and predicted activities) of model 1 (3b), model 2 (3d) and model 3 (3f)
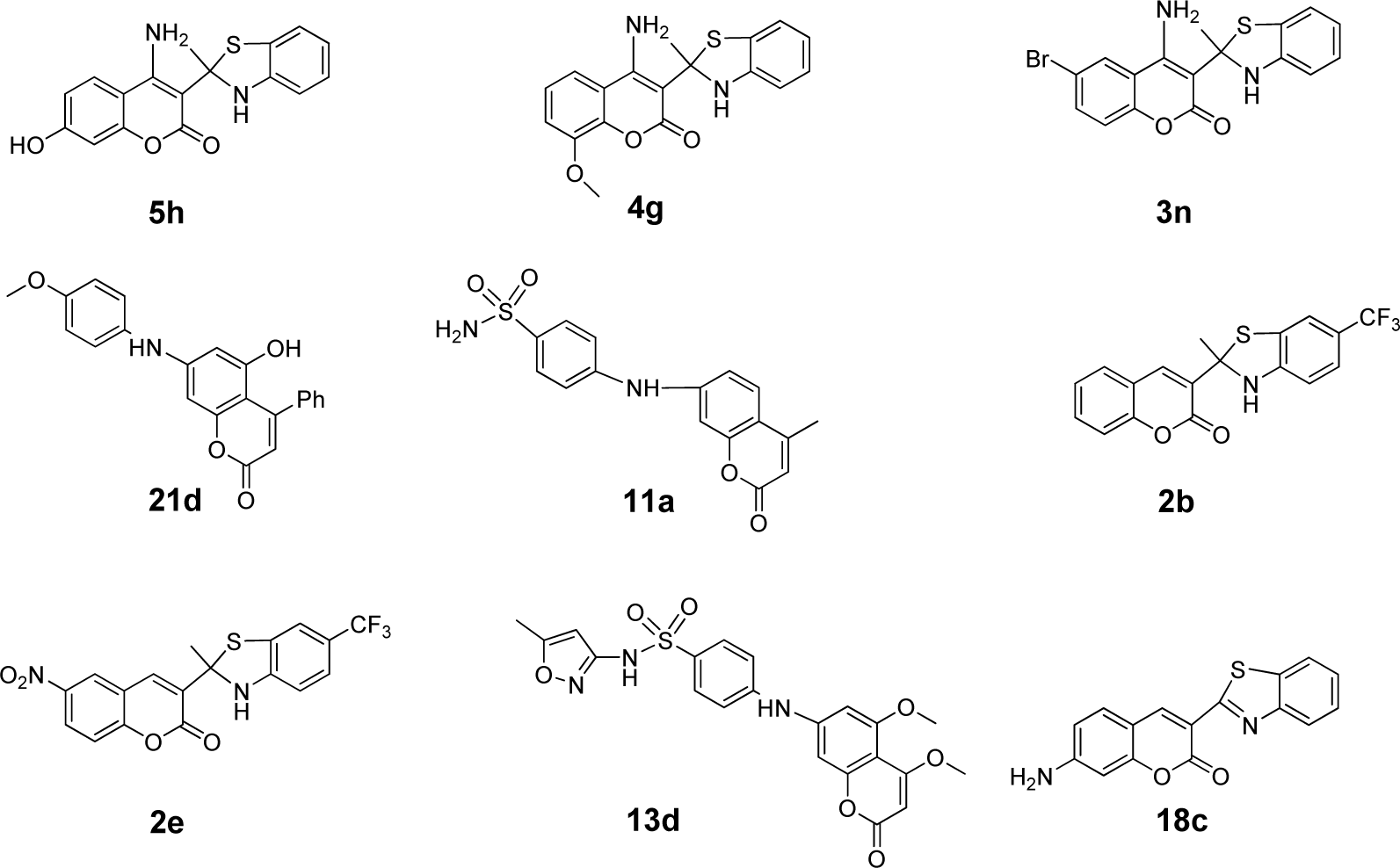
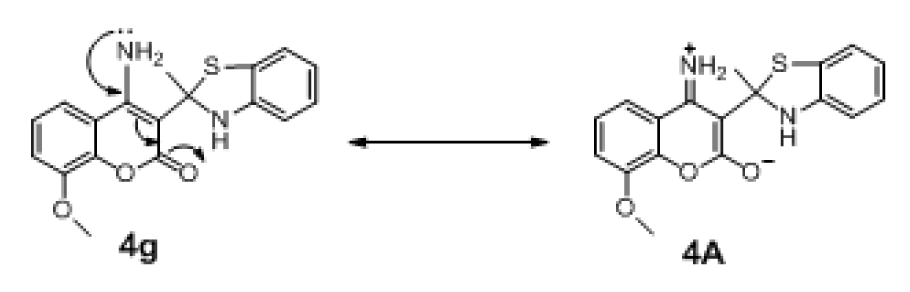
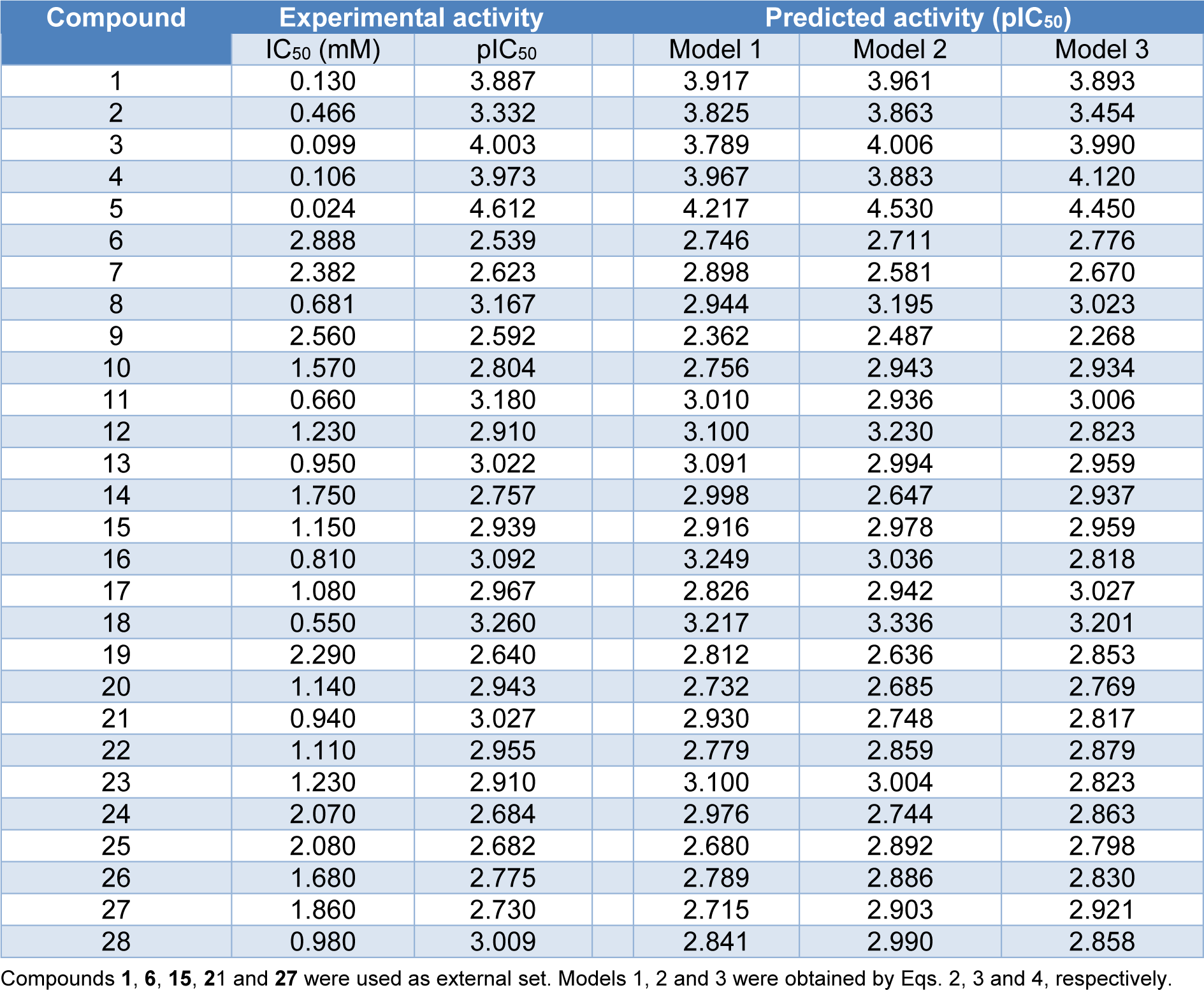
Table 1: Experimental and predicted antioxidant activities (pIC50) of coumarin derivatives (1-28) using multiple linear regression method
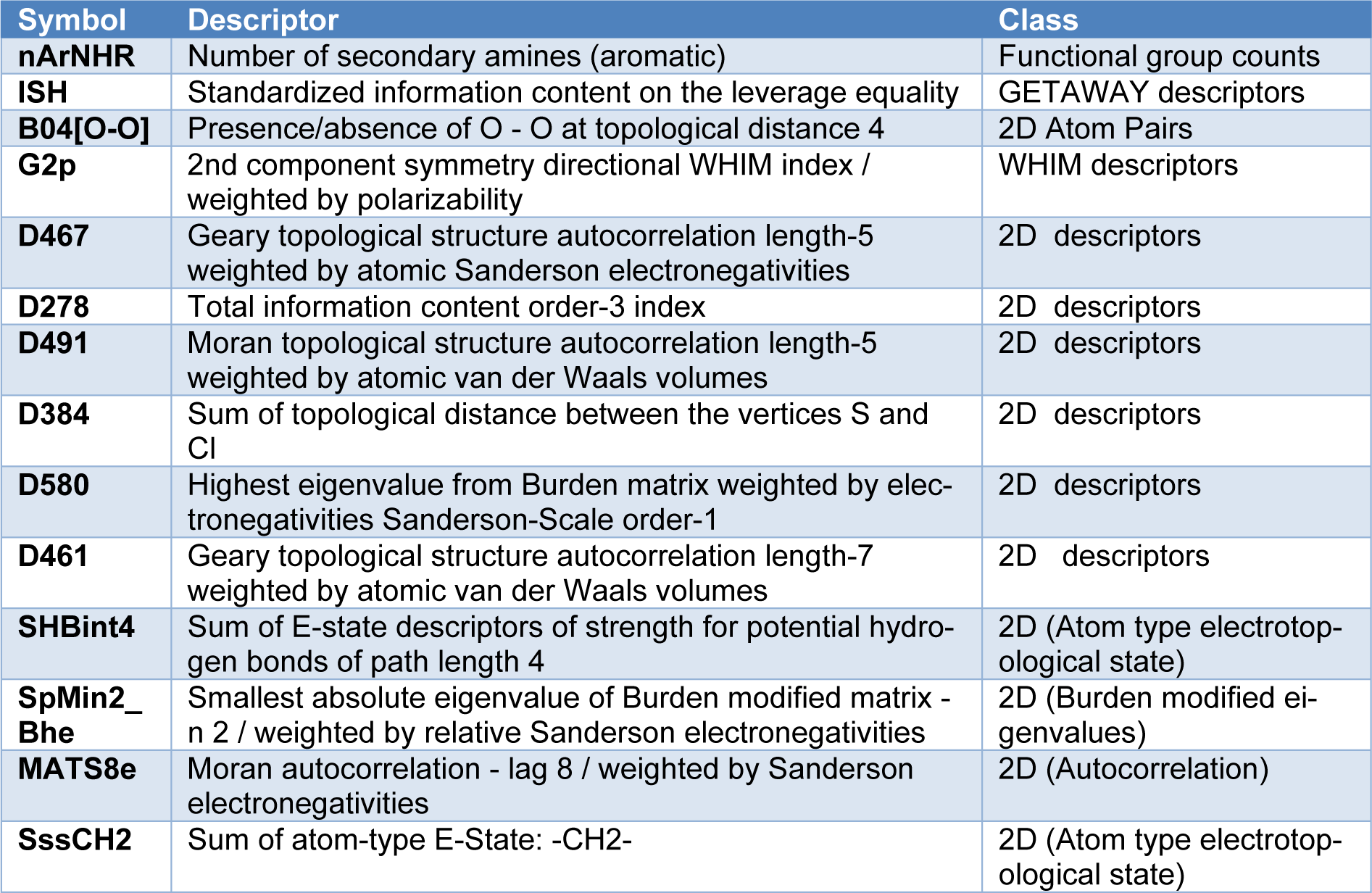
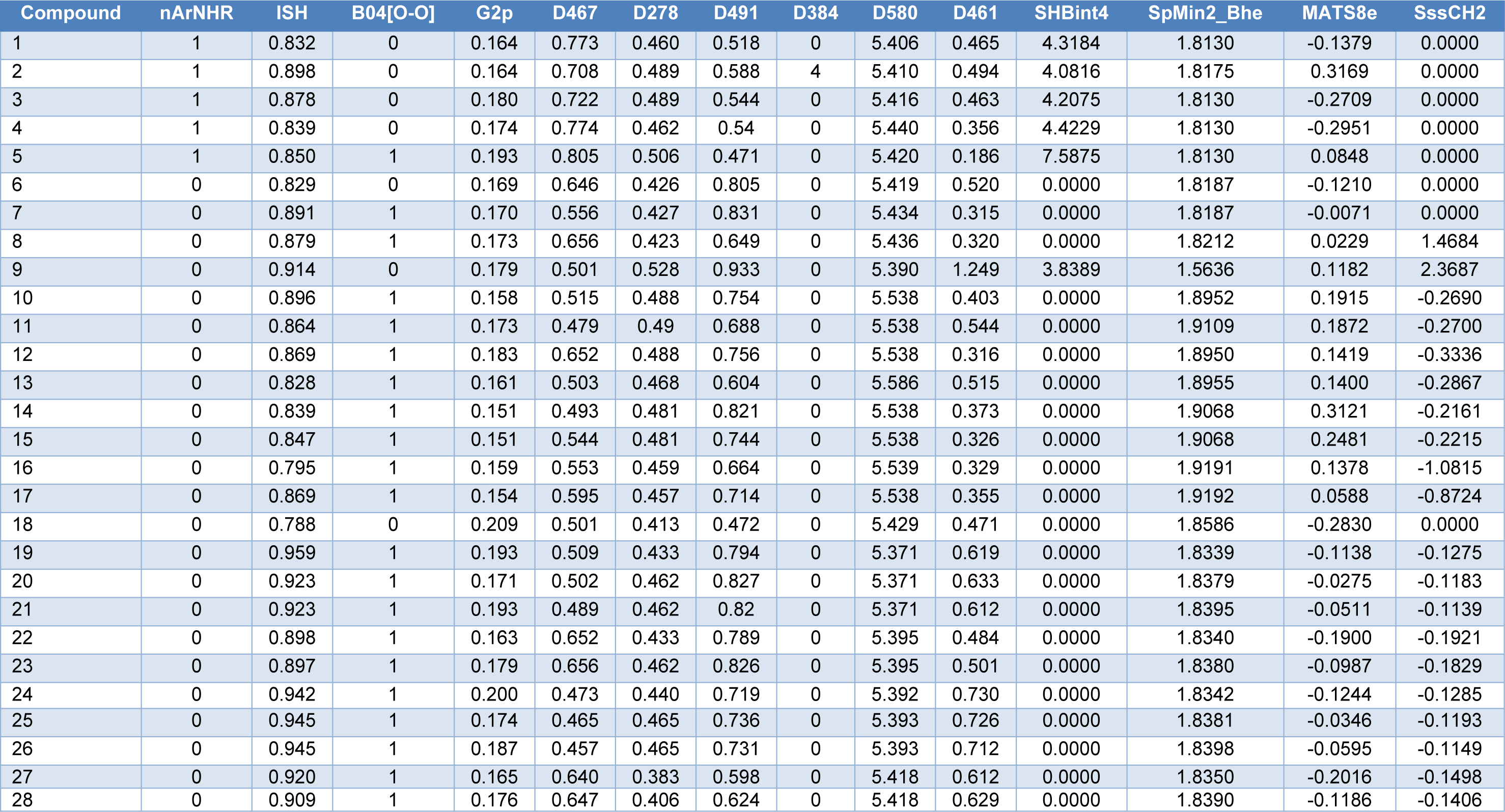
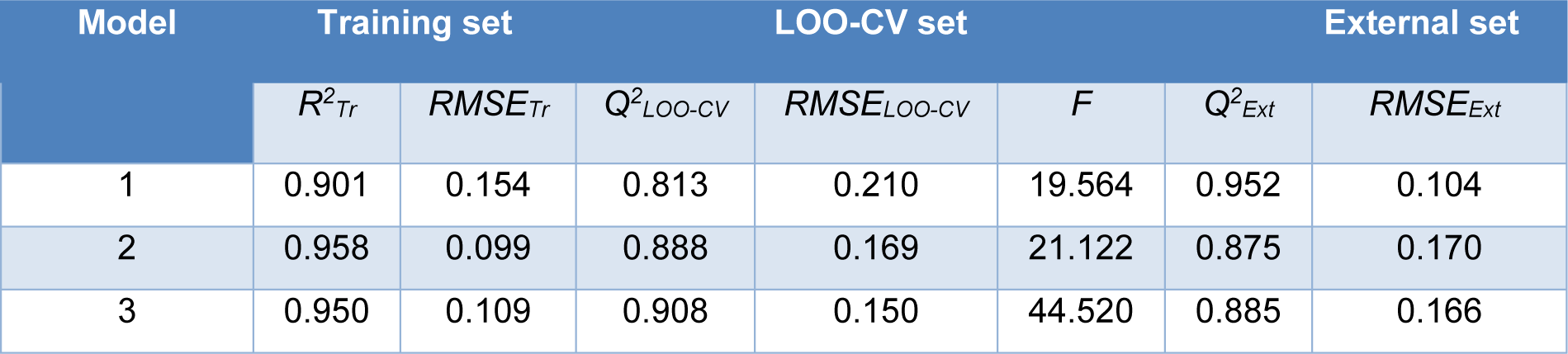
Table 4: Summary of statistical results in predicting antioxidant activity of coumarin derivatives (1-28)
[*] Corresponding Author:
Apilak Worachartcheewan, Department of Community Medical Technology, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand; Phone: (662) 441-4376, Fax: (662) 441-4380, eMail: apilak.woa@mahidol.ac.th