Letter to the editor
Mitigation of metabolic dyshomeostasis by glucocorticoid-receptor antagonism: Insights from animal and human studies
Aishwariya Madhavan1, Kusuma Murali1, Vaishnavi Raghavendra1, Apurva Kumar Ramesh Joshi1
1Department of Biochemistry, School of Sciences, Jain (Deemed to be University), Jayanagar 3rd Block, Bangalore, Karnataka, India 560041
EXCLI J 2020;19:Doc1266
Dear Editor,
Glucocorticoid hormones are steroidal signaling molecules produced by cortex of the adrenal gland. While acute glucocorticoid response is critical for immunomodulation and metabolic homeostasis, chronic elevated glucocorticoid levels have been recognized as a risk factor for metabolic syndrome (Wang, 2005[55]). Much of the understanding of consequences of excess glucocorticoids on metabolic homeostasis has come from observations on pathology associated with Cushing's syndrome. While incidence of Cushing syndrome is extremely low (Lindholm et al., 2001[35]), chronic exposure to excess glucocorticoids is a more realistic issue and needs to be taken into consideration. It is now well recognized that abnormalities such as diabetes/impaired glucose tolerance, obesity, hypertension and dyslipidemia are highly prevalent among patients of Cushing's syndrome (Chanson and Salenave, 2010[9]). A cross sectional study involving Cushing's syndrome patients clearly establishes the correlation between endogenous hypercortisolemia and metabolic abnormalities. The said study demonstrates that prompt diabetes was evident in 38 % of patients and fasting blood glucose, oral glucose tolerance test area under the curve (AUC) and HbA1C levels positively correlating with urinary free cortisol (Friedman et al., 1996[19]). The effect of glucocorticoids on metabolic homeostasis can also be discerned by the analysis of effects of corticosteroid therapy on glycemic regulations. Synthetic corticosteroids are the choice of drugs to treat various health issues such as asthma, chronic pulmonary obstructive disorders and rheumatoid arthritis. Panthakalam et al. (2004[40]) reported that 9 of 102 patients receiving glucocorticoid therapy for rheumatoid arthritis developed diabetes while pre-existing state of diabetes in another 6 worsened during the course of treatment (Panthakalam et al., 2004[40]). A retrospective analysis of medical data of patients who were hospitalized at general service of a hospital revealed that 64 % of patients receiving exogenous corticosteroid for at least 2 days developed hyperglycemia. This study demonstrates high prevalence of hyperglycemia among those receiving corticosteroid therapy and indicates that people with a history of diabetes before corticosteroid treatment are likely to develop hyperglycemia on corticosteroid therapy or due to other multiple co-morbidities (Donihi et al., 2006[13]).
Indeed, many experimental and clinical studies lend strong support to the view that excess glucocorticoid levels share causal relationship with various components of metabolic syndrome. Brunner et al. studied changes in autonomic cardiac activity and neuroendocrine functions in metabolic syndrome patients (n=30 vs. 153 control) of Whitehall II cohort. Interestingly, they observed that excretion (24 h) of a cortisol metabolite and normetanephrine increased in patients, in addition to higher levels of circulating interleukin-6 and C-reactive peptide (Brunner et al., 2002[7]). Analysis of cross-sectional data from the Paris Prospective Study revealed strong association of high systolic blood pressure with cortisol, blood glucose, heart rate, and free fatty acids (Filipovský et al., 1996[16]). A study conducted with young overweight Latino subjects revealed increased cortisol and fasting insulin levels in addition to increased 2 h glucose and insulin (during OGTT) levels among youth with metabolic syndrome. Further, systolic and diastolic blood pressure, fasting glucose levels and intra-abdominal fat tissue mass in subjects with MS were reported to correlate with cortisol levels, indicating that excess cortisol may have far reaching consequences on metabolic homeostasis (Weigensberg et al., 2008[58]). Similarly, another study conducted in obese children and adolescents revealed that circulating ACTH and cortisol levels were higher in metabolic syndrome subjects, who also had higher fasting glucose and insulin, increased systolic and diastolic blood pressure, and increased triglyceride levels (Sen et al., 2008[50]). To summarize, many human-subject based studies demonstrate that the excess glucocorticoid level is associated with many defining components of metabolic syndrome viz., increased waist circumference (Pasquali and Vicennati 2000[41]), increased triglyceride levels (Friedman et al., 1996[19]; Phillips et al., 1998[43]; Ward et al., 2003[56]), hypertension, increased blood glucose (Brunner et al., 2002[7]; Weigensberg et al., 2008[58]; Sen et al., 2008[50]) and insulin resistance (Phillips et al., 1998[43]; Ward et al., 2003[56]; Reinehr and Andler, 2004[47]).
Human subject-based studies clearly establish the association between cortisol and various metabolic aberrations associated with metabolic syndrome. While they offer clear perspectives on these correlations, much of the understanding of mechanisms responsible for the diabetogenic effects of glucocorticoids come from preclinical studies involving in vitro systems and experimental animal models. Owing to the fact that GCs are associated with insulin resistance, many authors have investigated the direct effect of GCs on beta cell functions employing isolated islets or insulin producing cell lines. Direct exposure to GCs appears to inhibit insulin release in vitro (Barseghian and Levine, 1980[2]; Gremlich et al., 1997[23]; Lambillotte et al., 1997[31]; Jeong et al., 2001[29]; Shinozuka et al., 2001[51]; Ullrich et al., 2005[54]; Zawalich et al., 2006[62]), an outcome which appears to be mediated by post-translational degradation of Glut2 protein (Gremlich et al., 1997[23]). Interestingly, this inhibitory effect is abolished by mifepristone, a glucocorticoid-antagonist, indicating involvement of receptor mediated mechanisms (Lambillotte et al., 1997[31]; Zawalich et al., 2006[62]). Despite compelling in vitro evidences for antagonistic effects of GCs on various aspects of beta cell functioning, such results were difficult to reproduce in vivo. On the contrary, paradoxically enough, experimental (Haber and Weinstein, 1992[24]; Giorgino et al., 1993[22]; Weinstein et al., 1993[59]; Holland et al., 2007[27]; Rafacho et al., 2009[46]; Protzek et al., 2014[45]) and human studies (Beard et al., 1984[3]; Willi et al., 2002[61]; Nicod et al., 2003[36]; Binnert et al., 2004[6]) demonstrate that administration of glucocorticoids results in hyperinsulinemia as a result of augmented beta cell function to compensate for peripheral insulin resistance.
Glucocorticoids are known for transcriptional activation genes of gluconeogenesis enzymes like G6Pase (Argaud et al., 1996[1]), phosphoenolpyruvate carboxykinase (PEPCK) (O'Brien et al., 1990[38]; Hanson and Reshef 1997[25]) and tyrosine aminotransferase (TAT) (Schmid et al., 1987[49]; Ganss et al., 1994[20]). In addition, GCs also facilitate muscle protein breakdown and increase the supply of amino acids that serve as gluconeogenesis substrates (Lecker et al., 1999[33]). Using the strategy of subjecting diabetic (insulin dependent) rats to adrenalectomy and glucocorticoid treatment, Exton et al., were able to demonstrate that hepatic glucose output was a consequence of glucocorticoid-dependent gluconeogenesis and was found to be underlined by up-regulation of PEPCK (Exton et al., 1973[14]). Glucocorticoids have been reported to have distinct effects on key mediators involved in the insulin signaling pathway. Cortisone treatment, which caused increase in blood glucose and insulin, was reported to be associated with reduced phosphorylation of insulin receptor without changes in the total levels of it, as well as reduced levels of IRS1 in skeletal muscle (Giorgino et al., 1993[22]). Saad et al., observed that dexamethasone reduced stimulated insulin receptor phosphorylation status in livers of rats, along with reduced phosphorylation of IRS1 and PI3K activity associated with IRS1. Further, dexamethasone was also found to be associated with reduced IRS1-associated PI3K activity in muscle as well (Saad et al., 1993[48]). The glucose transporter, Glut4 is the main insulin-responsive transporter that mediates insulin-induced glucose uptake in skeletal muscle and adipose tissue. The insulin-responsiveness of glut4 is characterized by insulin-induced translocation of the transporter to the plasma membrane from intracellular locations. Short-term treatment of rats with dexamethasone resulted in decrease in insulin stimulated glucose uptake, which was found to be underlined by impairments in cell surface recruitment of glut4 to the plasma membrane (Weinstein et al., 1998[60]).
With extensive research done, it is now apparent that excessive activation of glucocorticoid receptor plays a crucial role in the development of metabolic syndrome/T2D. Therefore glucocorticoid receptor antagonism may offer a viable approach for mitigating abnormalities associated with metabolic syndrome. This review intends to present an account of studies conducted with experimental animals as well as in human subjects that demonstrate efficacy of glucocorticoid antagonism in mitigating metabolic abnormalities typically associated with the metabolic syndrome.
The data on efficacy of GR antagonism in mitigating metabolic abnormalities is not limited to only preclinical studies. Human-subject based studies demonstrate that GR antagonism holds realistic promise in reducing the burden of metabolic abnormalities associated with excess glucocorticoids. It is important to recognize that much of our understanding of effect of mifepristone on glucocorticoid-related abnormalities has come from treatment of patients with Cushing's syndrome.
From studies conducted with various diabetic animal models, and studies conducted on Cushing's patients, it appears that GR antagonists have the potential to mitigate metabolic abnormalities associated with diabetes/metabolic syndrome. Despite its observed efficacy in alleviating anomalies associated with Cushing's syndrome, mifepristone has the disadvantage of lack of specificity (due to progesterone antagonism) and is associated with hypokalemia as a result of counter-regulatory activation of HPA axis-induced hypercortisolism (Castinetti et al., 2010[8]). While mifepristone is of great value in treating severe cases of Cushing's syndrome, selective GR antagonists that are devoid of propensity to cause activation of HPA axis are likely to be evaluated more for the possibility of therapeutic management of metabolic dysregulations associated with metabolic syndrome.
See also Table 1(Tab. 1) and 2(Tab. 2) (References in Table 1: Clapham and Turner, 1997[11]; Friedman et al., 1997[18]; Gettys et al., 1997[21]; Hashimoto et al., 2013[26]; Jacobson et al., 2005[28]; Kusunoki et al., 1995[30]; Langley and York, 1990[32]; Liang et al., 2005[34]; Okada et al.,1992[39]; Priyadarshini and Anuradha, 2017[44]; Takeshita et al., 2015[52]; Taylor et al., 2009[53]; Watts et al., 2005[57]; References in Table 2: Beaufrère et al., 1987[4]; Bertagna et al., 1986[5]; Chu et al., 2001[10]; Debono et al., 2013[12]; Fein et al., 2015[15]; Fleseriu et al., 2012[17]; Nieman et al., 1985[37]).
Acknowledgements
Authors are thankful to Jain (Deemed to be University), Bangalore for the support.
Conflict of interest
None.
References
1.
Argaud D, Zhang Q, Pan W, Maitra S, Pilkis SJ, Lange AJ. Regulation of rat liver glucose-6-phosphatase gene expression in different nutritional and hormonal states: gene structure and 5’-flanking sequence. Diabetes. 1996;45:1563–71.2.
Barseghian G, Levine R. Effect of corticosterone on insulin and glucagon secretion by the isolated perfused rat pancreas. Endocrinology. 1980;106:547–52.3.
Beard JC, Halter JB, Best JD, Pfeifer MA, Porte D. Dexamethasone-induced insulin resistance enhances B cell responsiveness to glucose level in normal men. Am J Physiol. 1984;247:E592-6.4.
Beaufrère B, De Parscau L, Chatelain P, Morel Y, Aguercif M, François R. RU 486 administration in a child with Cushing’s syndrome. Lancet. 1987;330:217. 5.
Bertagna X, Bertagna C, Laudat MH, Husson JM, Girard F, Luton JP. Pituitary-adrenal response to the antiglucocorticoid action of ru 486 in Cushing’s syndrome. J Clin Endocrinol Metab. 1986;63:639–43.6.
Binnert C, Ruchat S, Nicod N, Tappy L. Dexamethasone-induced insulin resistance shows no gender difference in healthy humans. Diabetes Metab. 2004;30:321–6. 7.
Brunner EJ, Hemingway H, Walker BR, Page M, Clarke P, Juneja M, et al. Adrenocortical, autonomic, and inflammatory causes of the metabolic syndrome: nested case-control study. Circulation. 2002;106:2659–65.8.
Castinetti F, Conte-Devolx B, Brue T. Medical treatment of Cushing’s syndrome: Glucocorticoid receptor antagonists and mifepristone. Neuroendocrinology. 2010;92:125–30.9.
Chanson P, Salenave S. Metabolic syndrome in Cushing’s syndrome. Neuroendocrinology. 2010;92:96–101.10.
Chu JW, Matthias DF, Belanoff J, Schatzberg A, Hoffman AR, Feldman D. Successful long-term treatment of refractory Cushing’s disease with high-dose mifepristone (RU 486). J Clin Endocrinol Metab. 2001;86:3568–73.11.
Clapham JC, Turner NC. Effects of the Glucocorticoid ii receptor antagonist mifepristone on hypertension in the obese Zucker rat. J Pharmacol Exp Ther. 1997;282:1503–8.12.
Debono M, Chadarevian R, Eastell R, Ross RJ, Newell-Price J. Mifepristone reduces insulin resistance in patient volunteers with adrenal incidentalomas that secrete low levels of cortisol: A pilot study. PLoS One. 2013;8:e60984.13.
Donihi AC, Raval D, Saul M, Korytkowski MT, DeVita MA. Prevalence and predictors of corticosteroid-related hyperglycemia in hospitalized patients. Endocr Pract. 2006;12:358–62.14.
Exton JH, Harper SC, Tucker AL, Flagg JL, Park CR. Effects of adrenalectomy and glucocorticoid replacement on gluconeogenesis in perfused livers from diabetic rats. Biochim Biophys Acta. 1973;329:41–57.15.
Fein HG, Vaughan TB, Kushner H, Cram D, Nguyen D. Sustained weight loss in patients treated with mifepristone for Cushing’s syndrome: A follow-up analysis of the SEISMIC study and long-term extension. BMC Endocr Disord. 2015;15:63.16.
Filipovský J, Ducimetière P, Eschwège E, Richard JL, Rosselin G, Claude JR. The relationship of blood pressure with glucose, insulin, heart rate, free fatty acids and plasma cortisol levels according to degree of obesity in middle-aged men. J Hypertens. 1996;14:229–35.17.
Fleseriu M, Biller BMK, Findling JW, Molitch ME, Schteingart DE, Gross C. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing’s syndrome. J Clin Endocrinol Metab. 2012;97:2039–49.18.
Friedman JE, Sun Y, Ishizuka T, Farrell CJ, McCormack SE, Herron LM, et al. Phosphoenolpyruvate carboxykinase (GTP) gene transcription and hyperglycemia are regulated by glucocorticoids in genetically obese db/db transgenic mice. J Biol Chem. 1997;272:31475–81. 19.
Friedman TC, Mastorakos G, Newman TD, Mullen NM, Horton EG, Costello R, et al. Carbohydrate and lipid metabolism in endogenous hypercortisolism: shared features with metabolic syndrome X and NIDDM. Endocr J. 1996;43:645–55.20.
Ganss R, Weih F, Schütz G. The cyclic adenosine 3’,5’-monophosphate- and the glucocorticoid-dependent enhancers are targets for insulin repression of tyrosine aminotransferase gene transcription. Mol Endocrinol. 1994;8:895–903.21.
Gettys TW, Watson PM, Taylor IL, Collins S. RU-486 (Mifepristone) ameliorates diabetes but does not correct deficient β-adrenergic signalling in adipocytes from mature C57BL/6J-ob/ob mice. Int J Obes. 1997;21:865–73. 22.
Giorgino F, Almahfouz A, Goodyear LJ, Smith RJ. Glucocorticoid regulation of insulin receptor and substrate IRS-1 tyrosine phosphorylation in rat skeletal muscle in vivo. J Clin Invest. 1993;91:2020–30.23.
Gremlich S, Roduit R, Thorens B. Dexamethasone induces posttranslational degradation of GLUT2 and inhibition of insulin secretion in isolated pancreatic beta cells. Comparison with the effects of fatty acids. J Biol Chem. 1997;272:3216–22. 24.
Haber RS, Weinstein SP. Role of glucose transporters in glucocorticoid-induced insulin resistance. GLUT4 isoform in rat skeletal muscle is not decreased by dexamethasone. Diabetes. 1992;41:728–35.25.
Hanson RW, Reshef L. Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu Rev Biochem. 1997;66:581–611.26.
Hashimoto T, Igarashi J, Hasan AU, Ohmori K, Kohno M, Nagai Y, et al. Mifepristone promotes adiponectin production and improves insulin sensitivity in a mouse model of diet-induced-obesity. PLoS One. 2013;8(11):e79724. 27.
Holland WL, Brozinick JT, Wang L-P, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–79.28.
Jacobson PB, Von Geldern TW, Öhman L, Österland M, Wang J, Zinker B, et al. Hepatic glucocorticoid receptor antagonism is sufficient to reduce elevated hepatic glucose output and improve glucose control in animal models of type 2 diabetes. J Pharmacol Exp Ther. 2005;314:191–200. 29.
Jeong I-K, Oh S-H, Kim B-J, Chung J-H, Min Y-K, Lee M-S, et al. The effects of dexamethasone on insulin release and biosynthesis are dependent on the dose and duration of treatment. Diabetes Res Clin Pract. 2001;51:163–71. 30.
Kusunoki M, Cooney GJ, Hara T, Storlien LH. Amelioration of high-fat feeding-induced insulin resistance in skeletal muscle with the antiglucocorticoid RU486. Diabetes. 1995;44:718–20.31.
Lambillotte C, Gilon P, Henquin JC. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J Clin Invest. 1997;99:414–23.32.
Langley SC, York DA. Effects of antiglucocorticoid RU 486 on development of obesity in obese fa/fa Zucker rats. Am J Physiol Regul Integr Comp Physiol. 1990;259:R539-44.33.
Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr. 1999;129:227S-37S.34.
Liang Y, Osborne MC, Monia BP, Bhanot S, Watts LM, She P, et al. Antisense oligonucleotides targeted against glucocorticoid receptor reduce hepatic glucose production and ameliorate hyperglycemia in diabetic mice. Metabolism. 2005;54:848–55.35.
Lindholm J, Juul S, Jørgensen JO, Astrup J, Bjerre P, Feldt-Rasmussen U, et al. Incidence and late prognosis of Cushing’s syndrome: a population-based study. J Clin Endocrinol Metab. 2001;86:117–23.36.
Nicod N, Giusti V, Besse C, Tappy L. Metabolic adaptations to dexamethasone-induced insulin resistance in healthy volunteers. Obes Res. 2003;11:625–31.37.
Nieman LK, Chrousos GP, Kellner C, Spitz IM, Nisula BC, Cutler GB, et al. Successful treatment of Cushing’s syndrome with the glucocorticoid antagonist RU 486. J Clin Endocrinol Metab. 1985;61:536–40.38.
O’Brien RM, Lucas PC, Forest CD, Magnuson MA, Granner DK. Identification of a sequence in the PEPCK gene that mediates a negative effect of insulin on transcription. Science. 1990;249:533–7.39.
Okada S, York DA, Bray GA. Mifepristone (RU 486), a blocker of type II glucocorticoid and progestin receptors, reverses a dietary form of obesity. Am J Physiol Regul Integr Comp Physiol. 1992;262:R1106-10. 40.
Panthakalam S, Bhatnagar D, Klimiuk P. The prevalence and management of hyperglycaemia in patients with rheumatoid arthritis on corticosteroid therapy. Scott Med J. 2004;49:139–41.41.
Pasquali R, Vicennati V. Activity of the hypothalamic–pituitary–adrenal axis in different obesity phenotypes. Int J Obes. 2000;24:S47–9. 42.
Phillips DIW, Barker DJP, Fall CHD, Seckl JR, Whorwood CB, Wood PJ, et al. Elevated plasma cortisol concentrations: A link between low birth weight and the insulin resistance syndrome? J Clin Endocrinol Metab. 1998;83:757–60.43.
Phillips DIW, Barker DJP, Fall CHD, Seckl JR, Whorwood CB, Wood PJ, et al. Elevated plasma cortisol concentrations: A link between low birth weight and the insulin resistance syndrome? J Clin Endocrinol Metab. 1998;83:757–60.44.
Priyadarshini E, Anuradha CV. Glucocorticoid antagonism reduces insulin resistance and associated lipid abnormalities in high-fructose-fed mice. Can J Diabetes. 2017;41:41–51. 45.
Protzek AOP, Costa-Júnior JM, Rezende LF, Santos GJ, Araújo TG, Vettorazzi JF, et al. Augmented β-cell function and mass in glucocorticoid-treated rodents are associated with increased islet Ir-β /AKT/mTOR and decreased AMPK/ACC and AS160 signaling. Int J Endocrinol. 2014;2014:983453.46.
Rafacho A, Cestari TM, Taboga SR, Boschero AC, Bosqueiro JR. High doses of dexamethasone induce increased beta-cell proliferation in pancreatic rat islets. Am J Physiol Endocrinol Metab. 2009;296:E681-9.47.
Reinehr T, Andler W. Cortisol and its relation to insulin resistance before and after weight loss in obese children. Horm Res. 2004;62:107–12.48.
Saad MJA, Folli F, Kahn JA, Kahn CR. Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J Clin Invest. 1993;92:2065–72.49.
Schmid E, Schmid W, Jantzen M, Mayer D, Jastorff B, Schütz G. Transcription activation of the tyrosine aminotransferase gene by glucocorticoids and cAMP in primary hepatocytes. Eur J Biochem. 1987;165:499–506.50.
Sen Y, Aygun D, Yilmaz E, Ayar A. Children and adolescents with obesity and the metabolic syndrome have high circulating cortisol levels. Neuro Endocrinol Lett. 2008;29:141-5.51.
Shinozuka Y, Okada M, Oki T, Sagane K, Mizui Y, Tanaka I, et al. Altered expression of HES-1, BETA2/NeuroD, and PDX-1 is involved in impaired insulin synthesis induced by glucocorticoids in HIT-T15 cells. Biochem Biophys Res Commun. 2001;287:229–35.52.
Takeshita Y, Watanabe S, Hattori T, Nagasawa K, Matsuura N, Takahashi K, et al. Blockade of glucocorticoid receptors with RU486 attenuates cardiac damage and adipose tissue inflammation in a rat model of metabolic syndrome. Hypertens Res. 2015;38:741–50. 53.
Taylor AI, Frizzell N, McKillop AM, Flatt PR, Gault VA. Effect of RU486 on hepatic and adipocyte gene expression improves diabetes control in obesity-type 2 diabetes. Horm Metab Res. 2009;41:899–904.54.
Ullrich S, Berchtold S, Ranta F, Seebohm G, Henke G, Lupescu A, et al. Serum- and glucocorticoid-inducible kinase 1 (SGK1) mediates glucocorticoid-induced inhibition of insulin secretion. Diabetes. 2005;54:1090–9.55.
Wang M. The role of glucocorticoid action in the pathophysiology of the metabolic syndrome. Nutr Metab. 2005;2:356.
Ward AMV, Fall CHD, Stein CE, Kumaran K, Veena SR, Wood PJ, et al. Cortisol and the metabolic syndrome in South Asians. Clin Endocrinol (Oxf). 2003;58:500–5.57.
Watts LM, Manchem VP, Leedom TA, Rivard AL, McKay RA, Bao D, et al. Reduction of hepatic and adipose tissue glucocorticoid receptor expression with antisense oligonucleotides improves hyperglycemia and hyperlipidemia in diabetic rodents without causing systemic glucocorticoid antagonism. Diabetes. 2005;54:1846–53.58.
Weigensberg MJ, Toledo-Corral CM, Goran MI. Association between the metabolic syndrome and serum cortisol in overweight Latino youth. J Clin Endocrinol Metab. 2008;93:1372–8.59.
Weinstein SP, Holand A, O’Boyle E, Haber RS. Effects of thiazolidinediones on glucocorticoid-induced insulin resistance and GLUT4 glucose transporter expression in rat skeletal muscle. Metabolism. 1993;42:1365–9.60.
Weinstein SP, Wilson CM, Pritsker A, Cushman SW. Dexamethasone inhibits insulin-stimulated recruitment of GLUt4 to the cell surface in rat skeletal muscle. Metabolism. 1998;47:3–6.61.
Willi SM, Kennedy A, Wallace P, Ganaway E, Rogers NL, Garvey WT. Troglitazone antagonizes metabolic effects of glucocorticoids in humans: Effects on glucose tolerance, insulin sensitivity, suppression of free fatty acids, and leptin. Diabetes. 2002;51:2895–902.62.
Zawalich WS, Tesz GJ, Yamazaki H, Zawalich KC, Philbrick W, Fajans SS, et al. Dexamethasone suppresses phospholipase C activation and insulin secretion from isolated rat islets. Metabolism. 2006;55:35–42.
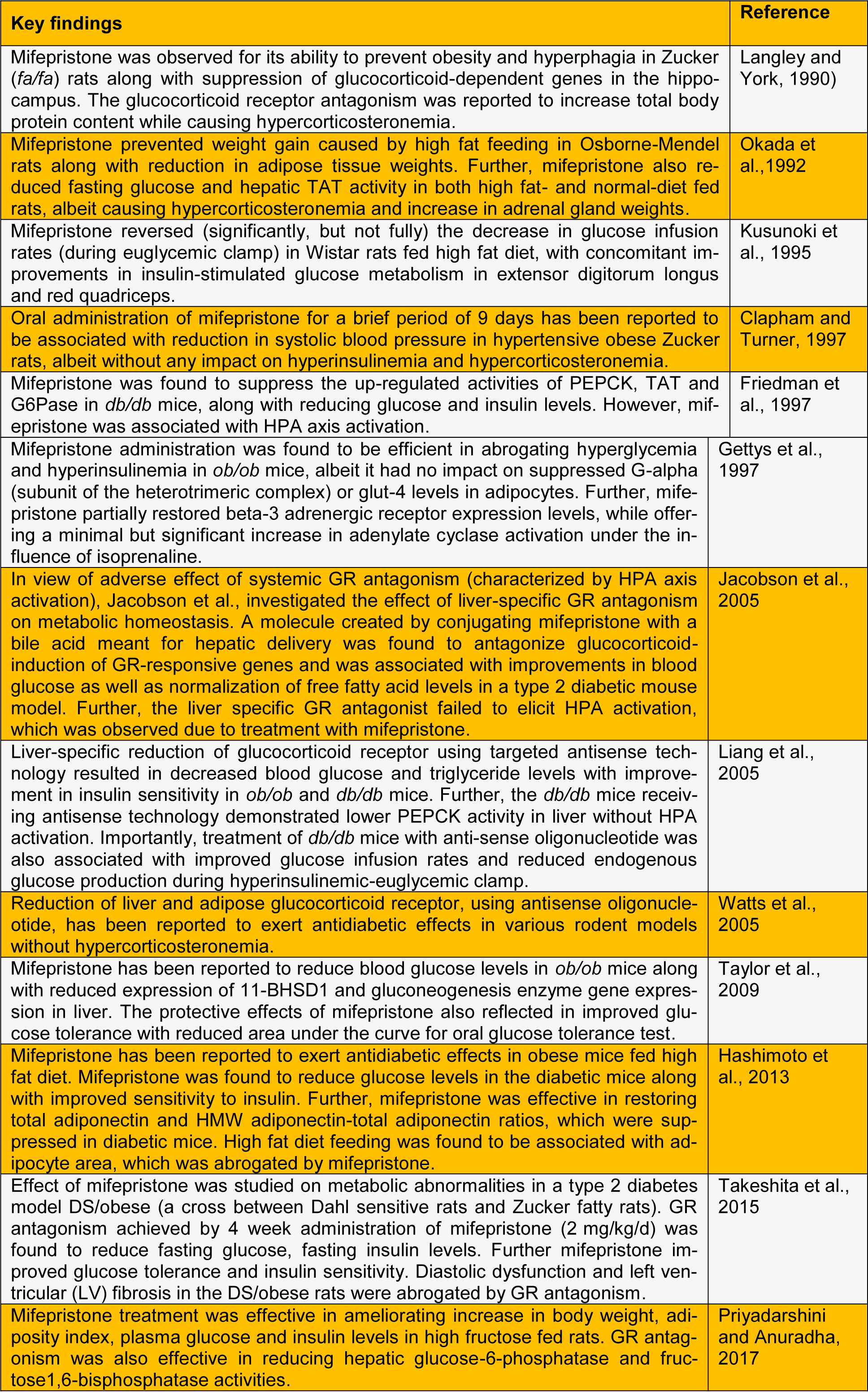
Table 1: Summary of experimental reports on the effect of glucocorticoid antagonism on metabolic aberrations
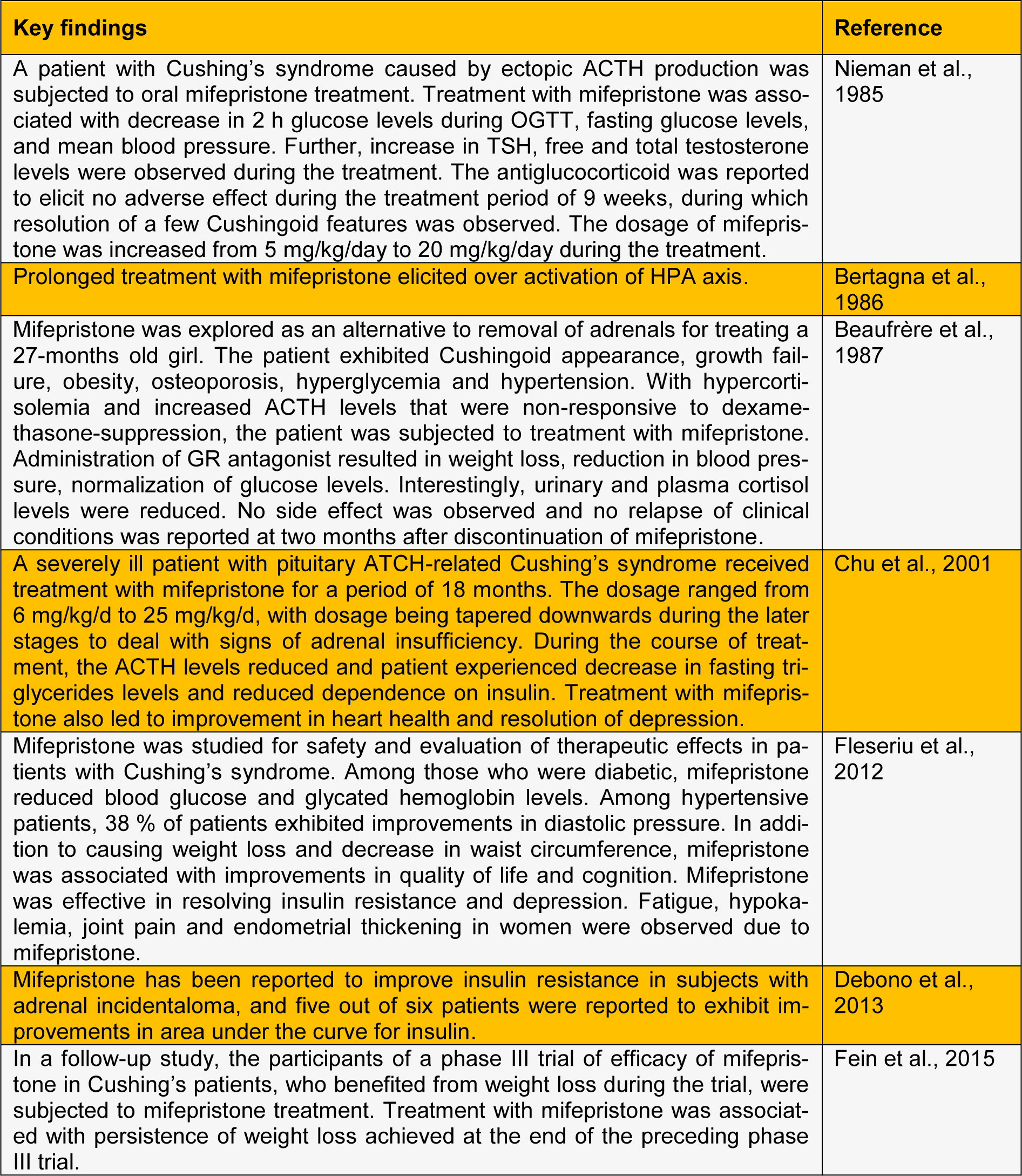
Table 2: Human-subject based studies demonstrating effect of glucocorticoid antagonism on metabolic aberrations