Review article
Diabetoporosis: Role of nitric oxide
Nasibeh Yousefzadeh1, Sajad Jeddi1, Khosrow Kashfi2,3, Asghar Ghasemi1
1Endocrine Physiology Research Center, Research Institute for Endocrine Sciences, Shahid Beheshti University of Medical Sciences, Tehran, Iran
2Department of Molecular, Cellular, and Biomedical Sciences, Sophie Davis School of Biomedical Education, City University of New York School of Medicine, NY, USA
3PhD Program in Biology, City University of New York Graduate Center, New York,NY, USA
EXCLI J 2021;20:Doc764
Abstract
Diabetoporosis, diabetic-related decreased bone quality and quantity, is one of the leading causes of osteoporotic fractures in subjects with type 2 diabetes (T2D). This is associated with lower trabecular and cortical bone quality, lower bone turnover rates, lower rates of bone healing, and abnormal posttranslational modifications of collagen. Decreased nitric oxide (NO) bioavailability has been reported within the bones of T2D patients and can be considered as one of the primary mechanisms by which diabetoporosis is manifested. NO donors increase trabecular and cortical bone quality, increase the rate of bone formation, accelerate the bone healing process, delay osteoporosis, and decrease osteoporotic fractures in T2D patients, suggesting the potential therapeutic implication of NO-based interventions. NO is produced in the osteoblast and osteoclast cells by three isoforms of NO synthase (NOS) enzymes. In this review, the roles of NO in bone remodeling in the normal and diabetic states are discussed. Also, the favorable effects of low physiological levels of NO produced by endothelial NOS (eNOS) versus detrimental effects of high pathological levels of NO produced by inducible NOS (iNOS) in diabetoporosis are summarized. Available data indicates decreased bone NO bioavailability in T2D and decreased expression of eNOS, and increased expression and activity of iNOS. NO donors can be considered novel therapeutic agents in diabetoporosis.
Keywords: Diabetoporosis, nitric oxide, diabetoporosis
Introduction
The prevalence of type 2 diabetes (T2D) is increasing worldwide and is estimated to reach 693 million by 2045 (Guariguata et al., 2014[50]; Cho et al., 2018[29]). Diabetoporosis that is diabetic-related changes in bone, characterized by decreased bone quality and quantity (Ferrari et al., 2018[41]), is one of the leading causes of osteoporotic fractures in subjects with T2D (Wongdee and Charoenphandhu, 2011[164]). A higher risk of osteoporotic fractures in T2D patients has been reported in several population-based studies (Forsén et al., 1999[42]; de Liefde et al., 2005[36]; Ahmed et al., 2006[3]; Lipscombe et al., 2007[94]). A meta-analysis of case-control and cohort studies from 1980 to 2016 indicates that the risk of osteoporotic fractures is 50-80 % higher in an individual with T2D (Janghorbani et al., 2007[72]; Vestergaard, 2007[151]; Moayeri et al., 2017[101]). In addition, it has been shown that osteoporotic fractures increase the 1-year mortality rate by 15-20 % in the elderly (Johnell and Kanis, 2004[75]; Wang et al., 2013[152]; González‐Zabaleta et al., 2016[49]). These data emphasize the need for developing new prevention/treatment strategies against osteoporotic fractures in patients with T2D.
Accumulating evidence indicates that decreased nitric oxide (NO) bioavailability can contribute to diabetoporosis. NO is produced in the osteoblast and osteoclast cells by the three isoforms of NO synthase (NOS) enzymes (Ralston et al., 1994[130]; Armour and Ralston, 1998[10]; Klein-Nulend et al., 1998[89]; Mancini et al., 2000[97]). In T2D, within the bone cells, the expression and activity of the endothelial NOS (eNOS) are decreased (Kalyanaraman et al., 2018[80]), while that of the inducible isoform, iNOS, is increased (MacPherson et al., 1999;[96] Bhatta et al., 2016[15]). eNOS-derived NO increases osteoblastic bone formation (Tai et al., 2007[142]; Jamal and Hamilton, 2012[71]) and directly inhibits osteoclast-mediated bone resorption (Wimalawansa, 2000[159][161]). In contrast, iNOS-derived NO inhibits osteoblast-mediated bone formation and a stimulatory effect on osteoclast-mediated bone resorption (Damoulis and Hauschka, 1997[35]; Hof and Ralston, 2001[61]; van't Hof et al., 2004[150]). It has been reported that eNOS deficiency decreases the rate of bone formation (Aguirre et al., 2001[2]; Armour et al., 2001[10]; Wimalawansa, 2009[156]), accelerates osteoporosis (Wimalawansa, 2010[158]), delays the bone healing process, and increases the risk of bone fractures (Hof and Ralston, 2001[61]; Jamal and Hamilton, 2012[71]). Also, it has been reported that NO donors have protective effects against osteoporotic bone fractures in postmenopausal women (Jamal et al., 2004[70]; Rejnmark et al., 2006[131]; Pouwels et al., 2010[122]) and in ovariectomized and corticosteroid-treated rats (Wimalawansa et al., 1996[163]; Samuels et al., 2001[136]). The role of NO on the function of the bone in the normal state has been previously reviewed (Kalyanaraman et al., 2016[78], 2018[80]). Here, we review the role of NO in diabetoporosis.
NO in the Bone
NO is produced in the cells of the bone by all three NOS isoforms, that is, eNOS, neural NOS (nNOS), and iNOS (Saura et al., 2010[138]). eNOS and nNOS are constitutively expressed and thus continuously produce low levels of NO. iNOS, on the other hand, is activated by certain stimuli, including proinflammatory cytokines, and produces high and biologically toxic concentrations of NO (Saura et al., 2010[138]). Effects of NO on bone function depend on its concentration (Joshua et al., 2014[76]), low physiological levels of NO have a stimulatory effect on normal bone formation (Ralston et al., 1995[129]; Wimalawansa, 2007[160]), development (Zaragoza et al., 2006[168]; Saura et al., 2010[138]), remodeling (Wimalawansa et al., 2000[157]), and fracture healing. In contrast, a pathologically high level of NO has inhibitory effects on all of these processes.
The eNOS gene is constitutively expressed in osteoblasts and osteocytes in both the fetus and adults (Helfrich et al., 1997[57]; Fox and Chow, 1998[44]). Furthermore, eNOS is also expressed in osteoclasts, bone marrow stromal cells, and chondrocytes of the epiphyseal growth plate (Mancini et al., 2000[97]; Wimalawansa, 2009[156]). Studies in rodents with targeted deletion of the eNOS gene have shown that eNOS-derived NO mediates the stimulatory effects of sex-steroid (Armour and Ralston, 1998[10]; Wimalawansa, 2010[158]), thyroid hormones (Kalyanaraman et al., 2014[81]), and mechanical loading on bone formation (Punjabi et al., 1992[125]; Fox et al., 1996[43]; Fox and Chow, 1998[44]). eNOS-deficient rodents show reduced prenatal and postnatal trabecular bone volume and cortical thickness, bone length, and bone mineral density (Armour et al., 2001[10]; Hefler et al., 2001[55]). In addition, eNOS-deficient mice have lower osteoblast (Afzal et al., 2004[1]) and higher osteoclast activities (Kasten et al., 1994[86]; Armour et al., 1999[10]; Percival et al., 1999[116]), thus presenting with a higher risk of osteoporotic fracture (Yan et al., 2010[165]) and lower rates of the bone healing process (Collin-Osdoby et al., 1995[30]) (Table 1(Tab. 1)).
In neonatal female rats, the iNOS gene was shown to be constitutively expressed in osteoblasts (Hukkanen et al., 1999[65]). However, under normal conditions, iNOS is not detectable in adults; pro-inflammatory cytokines induce its expression and activity in osteoclasts and pre-osteoclast cells (Brandi et al., 1995[19]; Zheng et al., 2006[171]; Wimalawansa, 2008[162]). iNOS-deficient mice do not have any apparent bone abnormalities during their adult life, but they have lower prenatal bone growth and bone length (Watanuki et al., 2002[154]) (Table 1(Tab. 1)). High concentrations of NO that is produced by iNOS inhibit the activity and proliferation of osteoblasts (Damoulis and Hauschka, 1997[35]; Hof and Ralston, 2001[61]; van't Hof et al., 2004[150]) and increases osteoclast activity in pathophysiological conditions (Mundy, 1993[105]; Chen et al., 2002[27], 2005[26]; Hao et al., 2005[54]; Ho et al., 2005[60]; Wimalawansa, 2008[162]; Rajfer et al., 2019[128]).
Some studies have failed to detect nNOS expression in the bone cells (Schmidt et al., 1992[139]; Helfrich et al., 1997[57]); however, nNOS expression has been reported in bone lining cells and in osteocytes (Helfrich et al., 1997[57]; Fox and Chow, 1998[44]) during skeletal development (Hukkanen et al., 1999[65]) and fracture healing (Zhu et al., 2001[172]). nNOS-deficient mice have higher trabecular and cortical bone mineral density and lower bone remodeling with lower numbers of osteoclasts and osteoblasts (Jung et al., 2003[77]; van't Hof et al., 2004[150]) (Table 1(Tab. 1)).
Diabetoporosis at a Glance
Despite having a normal or increased bone mineral density, T2D patients are at a higher risk of osteoporotic fractures (van Daele et al., 1995[149]; Sosa et al., 1996[140]; Bonds et al., 2006[16]). This paradox suggests that the etiology of osteoporotic fractures in T2D is different from that of the general population (Jindal et al., 2018[73]). According to a meta-analysis of association studies, the higher risk of osteoporotic fractures in T2D patients is associated with lower trabecular bone quality, that is incomplete, poorly connected, and widely spaced trabeculae (Ho-Pham and Nguyen, 2019[63]), and also with lower cortical bone quality, encompassing lower width and higher porosity (Patsch et al., 2013[115]). In addition, a lower bone turnover rate (Hygum et al., 2017[66]; Purnamasari et al., 2017[126]; Napoli et al., 2018[108]), a higher degree of mineralization (Pritchard et al., 2013[123]), a lower rate of bone healing (Norris and Parker, 2011[112]), and abnormal posttranslational modifications of collagen (Picke et al., 2019[120]) have been reported in diabetoporosis.
The pathophysiological mechanisms of diabetoporosis are quite complex but can be divided into direct and indirect effects (Palermo et al., 2017[114]). In addition to the direct effects of hyperglycemia and insulin resistance on bone quality (Figure 1(Fig. 1)), the increased risk of osteoporotic fractures may also be explained by the presence of diabetic complications, decreased physical activity, obesity, lower vitamin D levels, and a higher risk of falls (Oei et al., 2015[113]). Bone vasculature impairment, increased inflammation, oxidative stress (McFarlane et al., 2004[100]; Hofbauer et al., 2007[62]; Kalyanaraman et al., 2018[80]), and bone marrow adiposity (Costantini and Conte, 2019[32]) are key factors that contribute to higher incidences of osteoporotic fractures and delayed fracture healing in T2D (Figure 1(Fig. 1)).
As shown in Figure 1(Fig. 1), hyperglycemia and insulin resistance directly decrease osteoblast differentiation and activity by decreasing the expression of osteoblast-related markers, including the Runt-related transcription factor 2 (Runx-2), osteocalcin, bone morphogenetic protein-2 (BMP-2), osteopontin. Wnt signaling pathway is also suppressed (Inaba et al., 1995[68]; Chiu et al., 2004[28]; Mathieu et al., 2005[98]; Hamann et al., 2011[53]; Sarkar and Choudhury, 2013[137]; Lattanzio et al., 2014[90]; Perez-Diaz et al., 2015[117]; Wei et al., 2015[155]), osteoclast activation and differentiation are increased through increases in the expression of osteoclast-related markers including the nuclear factor of activated T cells (NFAT), receptor activator of nuclear factor-kappa-Β ligand (RANKL), and tartrate-resistant acid phosphatase (TRAP) (McFarlane et al., 2004[100]; Hofbauer et al., 2007[62]; Picke et al., 2016[121]; Kalyanaraman et al., 2018[80]). In addition, increasing serum concentrations of the Wnt inhibitors, sclerostin, and Dickkopf WNT signaling pathway inhibitor-1 (DKK-1) by osteocytes can decrease bone turnover rate in T2D. Hyperglycemia and insulin resistance also indirectly increase the expression of adipogenic markers such as peroxisome proliferator-activated receptor γ (PPAR-γ) in bones and have inhibitory effects on the activity and differentiation of osteoblasts by increasing fat accumulation in the marrow cavity of long bones. In addition, hyperglycemia and insulin resistance indirectly affect bone quality by increasing advanced glycation end products (AGEs), oxidative stress, inflammation, and impaired bone vasculature. These changes might explain the higher risk of bone fractures and osteoporosis and the lower rate of bone healing in T2D.
NO Bioavailability in Diabetic Bones
Decreased NO bioavailability has been reported in bones of humans and animals with T2D and can be considered as one of the main mechanisms in diabetoporosis. As shown in Figure 2(Fig. 2), lower eNOS expression (Kalyanaraman et al., 2018[79]) or activity (Mordwinkin et al., 2012[103]) resulting in diminished NO synthesis and increased NO oxidation due to NO quenching by AGEs (Bucala et al., 1991[20]; Alikhani et al., 2007[5]) are the leading causes of decreased NO bioavailability in the diabetic bone. In addition, reduced availability of L-arginine, the substrate for the NOS enzymes, increases arginase activity (Bhatta et al., 2016[15]); increased expression and activity of iNOS (MacPherson et al., 1999[96]), impaired vasculature of the bones (Stabley et al., 2015[141]), uncoupling of eNOS (Kalyanaraman et al., 2018[80]), and damaged to the eNOS-caveolin-1 complex (Aicher et al., 2003[4]; Cao et al., 2012[22]) may be involved in decreased NO bioavailability in the diabetic bones.
eNOS uncoupling in the bones of T2D patients is at least in part due to increased production of bone morphogenetic protein 4 (BMP4) (Youn et al., 2015[167]) that leads to an eNOS-mediated superoxide production (Thum et al., 2007[146]). Lower activity of the eNOS/cGMP/PKG pathway due to the uncoupling of eNOS, inhibition of guanylate cyclase activity, and suppression of PKG transcription have all been reported in diabetic bones (Kalyanaraman et al., 2018[80]). In T2D, endothelial progenitor cells synthesize less NO because of the damaged eNOS-caveolin-1 complex (Aicher et al., 2003[4]; Cao et al., 2012[22]) that is associated with increased serum levels of Dickkopf-1, which is an inhibitor of osteoblast differentiation (Lattanzio et al., 2014[90]).
Bone remodeling is a life-long process and is achieved within anatomical structures that are known as a basic multicellular unit (BMU). These provide a unique microenvironment to facilitate coupled bone resorption and formation (Andersen et al., 2009[7]; Raggatt and Partridge, 2010[127]). Bone remodeling has four consecutive steps including activation, resorption, reversal, and formation, which require a coordinated action of the bone cells, including osteocytes, osteoblasts, osteoclasts, and bone-lining cells (Feng and McDonald, 2011[40]).
Step 1 (activation), the osteocytes sense signals for initiating remodeling; these include mechanical forces, changes in calcium homeostasis, or changes in hormone levels that translate into biological signals (Bonewald, 2007[17]; Raggatt and Partridge, 2010[127]). In osteocytes, initiating bone remodeling signals inhibit the expression of transforming growth factor β (TGF-β, as an inhibitor of bone resorption) (Heino et al., 2002[56]; Raggatt and Partridge, 2010[127]), and with a delay of about 5 days inhibit the expression of sclerostin (SOST, an inhibitor of bone formation) (van Bezooijen et al., 2004[148]; Li et al., 2005[92]; Robling et al., 2008[132]; Gasser and Kneissel, 2017[46]). In addition, the bone lining cells create a raised canopy above the remodeling surface, which merges with the bone vasculature for recruitment of osteoclast and osteoblast progenitor cells to the BMU (Arias et al., 2018[8]).
Step 2 (resorption), the decreased TGF-β in osteocytes recruits hematopoietic stem cells (HSC) from the bone marrow or the circulation; these HCSs are then differentiated to osteoclasts in the presence of monocyte/macrophage colony-stimulating factor (M-CSF) and the RANKL (Boyle et al., 2003[18]). Low levels of TGF-β increase the RANKL/osteoprotegerin (OPG) ratio and M-CSF expression in preosteoblasts (Karst et al., 2004[85]). OPG negatively regulates RANKL binding to RANK that is essential for activation and differentiation of osteoclasts (Karst et al., 2004[85]). In this step, osteoclasts digest organic and inorganic bone matrices by secreting acid phosphatase, cathepsin K, and collagenase, a process known as bone resorption (Henriksen et al., 2011[58]).
Mononuclear macrophage-like cells in step 3 (reversal) engulf and remove demineralized undigested collagen and generate transition signals that stop bone resorption and start bone formation (Raggatt and Partridge, 2010[127]).
Step 4 (formation and mineralization), in response to a decrease in SOST within osteocytes, mesenchymal stem cells (MSC) are recruited and differentiated into osteoblasts that start the bone formation and mineralization process. When an equal quantity of resorbed bone has been replaced, the remodeling cycle is terminated (Franz‐Odendaal et al., 2006[45]). Some osteoblasts in this step undergo apoptosis, others turn into lining cells, still, others remain trapped within the bone matrix and become osteocytes (Figure 3(Fig. 3)).
In T2D and the bone remodeling process, there is a decrease in eNOS-derived NO and an increase in iNOS-derived NO; this leads to inhibition of steps 1 and 4, activation and bone formation, respectively; and at the same time, step 2, bone formation is stimulated. As shown in Figure 3(Fig. 3), T2D decreases the production of eNOS-derived NO in osteocytes and, therefore, decreases osteocytes' capabilities in detecting and initiating the bone remodeling signals (step 1) (Collin-Osdoby et al., 2000[31]; Bakker et al., 2009[14]). eNOS-derived NO increases in response to mechanical forces, thyroid hormones, and estrogens (Fox et al., 1996[43]; Armour and Ralston, 1998[10]; Kalyanaraman et al., 2014[81]). T2D by decreasing eNOS-derived NO and increasing iNOS-derived NO increases bone resorption (step 2). eNOS-derived NO in T2D inhibits the production of M-CSF and RANKL and stimulates the production of OPG in both preosteoblasts and osteoblasts; these effects result in a decrease in recruitment of HSC and their differentiation to osteoclast (Wongdee and Charoenphandhu, 2011[164]; Catalfamo et al., 2013[23]). In addition, eNOS-derived NO decreases the activities of cathepsin K, a marker of high bone resorption and collagenase in osteoclast (Percival et al., 1999[116]; Gyurko et al., 2005[51]; Alselami et al., 2015[6]). Therefore, T2D increases bone resorption by decreasing eNOS-derived NO (Pezhman et al., 2019[119]). T2D stimulates the production of iNOS-derived NO, which increases PPARγ production by HSC and, therefore, stimulates differentiation of HSC to osteoclasts; in addition, iNOS-derived NO increases the activities of cathepsin K and collagenase and osteoclast activity (Percival et al., 1999[116]; Gyurko et al., 2005[51]; Alselami et al., 2015[6]). These effects result in increased bone resorption.
eNOS-derived NO directly activates and facilitates osteoblastic differentiation from MSC (Hikiji et al., 1997[59]) through phosphorylation of JNK/MAPK in preosteoblasts (Yang et al., 2018[166]). After transportation to the nucleus, p-JNK induces the expression of osteogenic transcription factors such as Runx2, osterix (OSX), and osteopontin (OPN) (Aguirre et al., 2001[2]) and represses the expression of adipogenic transcription factors such as PPARγ and lipoprotein lipase (LPL), thus increasing osteogenesis and decreasing adipogenesis (Rosen et al., 1999[133]; Aguirre et al., 2001[2]; Zhao et al., 2016[170]; Yang et al., 2018[166]). In addition, eNOS-derived NO directly activates osteoblast activity by increasing the alkaline phosphatase (ALPase) (Inoue et al., 1995[69]) and osteocalcin levels (Pun et al., 1989[124]) as well as increasing intracellular concentrations of cGMP (Hagiwara et al., 1996[52]).
Treatment of Diabetoporosis by Nitric Oxide
Available treatments for osteoporosis are limited by cost, side effects, and efficacy, with limited impact on the cortical bone (Table 2(Tab. 2); References in Table 2: Austin and Heath, 1981[12]; Burr et al., 2015[21]; Chau and Edelman, 2002[24]; Chen et al., 2014[25]; Dai et al., 2020[33]; Fabre et al., 2020[38]; Gattereau et al., 1980[47]; Ilich and Kerstetter, 2000[67]; John et al., 2011[74]; Kanazawa, 2017[82]; Karras et al., 2018[84]; Khosla and Hofbauer, 2017[88]; Khosla et al., 2007[87]; Li et al., 2018[91]; Lim and Bolster, 2017[93]; Mauvais-Jarvis et al., 2013[99]; Mohsin et al., 2019[102]; Morita et al., 2016[104]; Neer et al., 2001[109]; Nemeth and Goodman, 2016[111]; Nemeth et al., 2001[110]; Pettway et al., 2008[118]; Russell, 2011[134]; Russo and Russo, 2006[135]; Takakura et al., 2017[143]; Tella and Gallagher, 2014[144]; Tella et al., 2017[145]; Tsourdi et al., 2017[147]; Ward and Rauch, 2018[153]; Zhang et al., 2020[169]). Therefore, there is a need for easily administered and inexpensive agents that increase bone trabecular and cortical strength and decrease the risk of osteoporotic fractures. NO donors have a high potential to be cost‐effective novel therapeutic agents against osteoporosis and, in particular, against diabetoporosis.
Organic nitrates are used for treating heart failure and hypertension; epidemiological studies have shown that their use can reduce the risk of osteoporotic fractures (Rejnmark et al., 2006[131]; Pouwels et al., 2010[122]). Based on these observations, the protective effects of organic nitrates against osteoporotic fractures were reported in ovariectomized and corticosteroid-treated rats (Wimalawansa et al., 1996[163]; Samuels et al., 2001[136]; Wimalawansa, 2007[160], 2009[156]), mice (Wimalawansa et al., 1996[163]; Hukkanen et al., 2003[64]), and in ovariectomized (Wimalawansa, 2000[159]; Nabhan and Rabie, 2008[107]) and postmenopausal women (Wimalawansa et al., 1996[163]). Organic nitrates stimulate osteoblast-mediated bone formation (Wimalawansa et al., 1996[163]; Wimalawansa, 2000[161]) and inhibit osteoclast-mediated bone resorption (Fan et al., 2004[39]), thus decreasing the risk of osteoporotic fractures. Organic nitrates are the only FDA-approved NO donors, but their potential benefits are rapidly lost on long-term use due to the possible development of tolerance and endothelial dysfunction (Daiber and Münzel, 2015[34]). Inorganic nitrites and nitrates are NO donors with strong NO-like effects in both animals and humans; it has been suggested that they can act as suitable alternatives to organic nitrates (Münzel and Daiber, 2018[106]). These agents can protect against diabetoporosis directly by decreasing osteoclast activity and increasing osteoblast activity (see section “NO in the bone”), or indirectly, by improving the metabolic status (Ghasemi and Jeddi, 2017[48]; Lundberg et al., 2018[95]; Kapil et al., 2020[83]) and decreasing body weight (Bahadoran et al., 2020[13]).
Conclusion and Future Perspective
Decreased bone NO bioavailability in T2D is one of the primary mechanisms underlying diabetoporosis. This reduced NO bioavailability is due to decreased expression of eNOS, availability of L-arginine, and activity of cGMP/PKG, as well as increased eNOS uncoupling, expression, and activity of iNOS and arginase. NO donors can potentially be used as safe and cost‐effective novel therapeutic agents in diabetoporosis. This issue, however, remains to be verified in a well-designed clinical trial.
Notes
Khosrow Kashfi and Asghar Ghasemi (Endocrine Physiology Research Center, Research Institute for Endocrine Sciences, Shahid Beheshti University of Medical Sciences, No. 24, Parvaneh Street, Velenjak, P.O. Box: 19395-4763, Tehran, Iran; E-mail: Ghasemi@endocrine.ac.ir) contributed equally as corresponding author.
Acknowledgements
This study was supported by Shahid Beheshti University of Medical Sciences [grant No. 27443-1], Tehran, Iran.
Conflict of interest
The authors declare that they have no competing interests.
References
1.
Afzal F, Polak J, Buttery L. Endothelial nitric oxide synthase in the control of osteoblastic mineralizing activity and bone integrity. J Pathol. 2004;202:503-10.2.
Aguirre J, Buttery L, O'Shaughnessy M, Afzal F, Fernandez de Marticorena I, Hukkanen M, et al. Endothelial nitric oxide synthase gene-deficient mice demonstrate marked retardation in postnatal bone formation, reduced bone volume, and defects in osteoblast maturation and activity. Am J Pathol. 2001;158:247-57.3.
Ahmed LA, Joakimsen RM, Berntsen GK, Fønnebø V, Schirmer H. Diabetes mellitus and the risk of non-vertebral fractures: The Tromsø study. Osteoporos Int. 2006;17:495-500.4.
Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370-6.5.
Alikhani M, Alikhani Z, Boyd C, MacLellan CM, Raptis M, Liu R, et al. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone. 2007;40:345-53.6.
Alselami NM, Noureldeen AF, Al-Ghamdi MA, Khan JA, Moselhy SS. Bone turnover biomarkers in obese postmenopausal Saudi women with type-II diabetes mellitus. Afr Health Sci. 2015;15:90-6.7.
Andersen TL, Sondergaard TE, Skorzynska KE, Dagnaes-Hansen F, Plesner TL, Hauge EM, et al. A physical mechanism for coupling bone resorption and formation in adult human bone. Am J Pathol. 2009;174:239-47.8.
Arias CF, Herrero MA, Echeverri LF, Oleaga GE, López JM. Bone remodeling: A tissue-level process emerging from cell-level molecular algorithms. PloS One. 2018;13:e0204171.9.
Armour KE, Armour KJ, Gallagher ME, Gödecke A, Helfrich MH, Reid DM, et al. Defective bone formation and anabolic response to exogenous estrogen in mice with targeted disruption of endothelial nitric oxide synthase. Endocrinology. 2001;142:760-6.10.
Armour KE, Ralston SH. Estrogen upregulates endothelial constitutive nitric oxide synthase expression in human osteoblast-like cells. Endocrinology. 1998;139:799-802.11.
Armour KE, Van THRJ, Grabowski PS, Reid DM, Ralston SH. Evidence for a pathogenic role of nitric oxide in inflammation-induced osteoporosis. J Bone Mineral Res. 1999;14:2137-42.12.
Austin LA, Heath H 3rd. Calcitonin: Physiology and pathophysiology. N Engl J Med. 1981;304:269-78.13.
Bahadoran Z, Jeddi S, Gheibi S, Mirmiran P, Kashfi K, Ghasemi A. Inorganic nitrate, a natural anti-obesity agent: A systematic review and meta-analysis of animal studies. EXCLI J. 2020;19:972-83.14.
Bakker AD, Silva VC, Krishnan R, Bacabac RG, Blaauboer ME, Lin YC, et al. Tumor necrosis factor alpha and interleukin-1beta modulate calcium and nitric oxide signaling in mechanically stimulated osteocytes. Arthritis Rheum. 2009;60:3336-45.15.
Bhatta A, Sangani R, Kolhe R, Toque HA, Cain M, Wong A, et al. Deregulation of arginase induces bone complications in high-fat/high-sucrose diet diabetic mouse model. Mol Cell Endocrinol. 2016;422:211-20.16.
Bonds DE, Larson JC, Schwartz AV, Strotmeyer ES, Robbins J, Rodriguez BL, et al. Risk of fracture in women with type 2 diabetes: The Women's Health Initiative Observational Study. J Clin Endocrinol Metab. 2006;91:3404-10.17.
Bonewald LF. Osteocytes as dynamic multifunctional cells. Ann N Y Acad Sci. 2007;1116:281-90.18.
Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337-42.19.
Brandi ML, Hukkanen M, Umeda T, Moradi-Bidhendi N, Bianchi S, Gross SS, et al. Bidirectional regulation of osteoclast function by nitric oxide synthase isoforms. Proc Natl Acad Scie U S A. 1995;92:2954-8.20.
Bucala R, Tracey KJ, Cerami A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J Clin Invest. 1991;87:432-8.21.
Burr DB, Liu Z, Allen MR. Duration-dependent effects of clinically relevant oral alendronate doses on cortical bone toughness in beagle dogs. Bone. 2015;71:58-62.22.
Cao C, Zhang H, Gong L, He Y, Zhang N. High glucose conditions suppress the function of bone marrow-derived endothelial progenitor cells via inhibition of the eNOS-caveolin-1 complex. Mol Med Rep. 2012;5:341-6.23.
Catalfamo DL, Britten TM, Storch DL, Calderon NL, Sorenson HL, Wallet SM. Hyperglycemia induced and intrinsic alterations in type 2 diabetes-derived osteoclast function. Oral Dis. 2013;19:303-12.24.
Chau DL, Edelman SV. Osteoporosis and diabetes. Clin Diabetes. 2002;20:153.25.
Chen F, Ouyang Y, Ye T, Ni B, Chen A. Estrogen inhibits RANKL-induced osteoclastic differentiation by increasing the expression of TRPV5 channel. J Cell Biochem. 2014;115:651-8.26.
Chen RM, Chen TL, Chiu WT, Chang CC. Molecular mechanism of nitric oxide-induced osteoblast apoptosis. J Orthop Res. 2005;23:462-8.27.
Chen RM, Liu HC, Lin YL, Jean WC, Chen JS, Wang JH. Nitric oxide induces osteoblast apoptosis through the de novo synthesis of Bax protein. J Orthop Res. 2002;20:295-302.28.
Chiu KC, Chu A, Go VL, Saad MF. Hypovitaminosis D is associated with insulin resistance and beta cell dysfunction. Am J Clin Nutr. 2004;79:820-5.29.
Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, et al. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. 2018;138:271-81.30.
Collin-Osdoby P, Nickols GA, Osdoby P. Bone cell function, regulation, and communication: a role for nitric oxide. J Cell Biochem. 1995;57:399-408.31.
Collin-Osdoby P, Rothe L, Bekker S, Anderson F, Osdoby P. Decreased nitric oxide levels stimulate osteoclastogenesis and bone resorption both in vitro and in vivo on the chick chorioallantoic membrane in association with neoangiogenesis. J Bone Mineral Res. 2000;15:474-88.32.
Costantini S, Conte C. Bone health in diabetes and prediabetes. World J Diabetes. 2019;10:421-45.33.
Dai R, Wu Z, Chu HY, Lu J, Lyu A, Liu J, et al. Cathepsin K: The action in and beyond bone. Front Cell Dev Biol. 2020;8:433.34.
Daiber A, Münzel T. Organic nitrate therapy, nitrate tolerance, and nitrate-induced endothelial dysfunction: Emphasis on redox biology and oxidative stress. Antioxid Redox Signal. 2015;23:899-942.35.
Damoulis PD, Hauschka PV. Nitric oxide acts in conjunction with proinflammatory cytokines to promote cell death in osteoblasts. J Bone Mineral Res. 1997;12:412-22.36.
de Liefde II, van der Klift M, de Laet CE, van Daele PL, Hofman A, Pols HA. Bone mineral density and fracture risk in type-2 diabetes mellitus: The Rotterdam Study. Osteoporos Int. 2005;16:1713-20.37.
Diwan AD, Wang MX, Jang D, Zhu W, Murrell GA. Nitric oxide modulates fracture healing. J Bone Mineral Res. 2000;15:342-51.38.
Fabre S, Funck-Brentano T, Cohen-Solal M. Anti-sclerostin antibodies in osteoporosis and other bone diseases. J Clin Med. 2020;9(11):3439.39.
Fan X, Roy E, Zhu L, Murphy TC, Ackert-Bicknell C, Hart CM, et al. Nitric oxide regulates receptor activator of nuclear factor-kappaB ligand and osteoprotegerin expression in bone marrow stromal cells. Endocrinology. 2004;145:751-9.40.
Feng X, McDonald JM. Disorders of bone remodeling. Annu Rev Pathol. 2011;6:121-45.41.
Ferrari SL, Abrahamsen B, Napoli N, Akesson K, Chandran M, Eastell R, et al. Diagnosis and management of bone fragility in diabetes: an emerging challenge. Osteoporos Int. 2018;29:2585-96.42.
Forsén L, Meyer HE, Midthjell K, Edna TH. Diabetes mellitus and the incidence of hip fracture: Results from the Nord-Trøndelag Health Survey. Diabetologia. 1999;42:920-5.43.
Fox SW, Chambers TJ, Chow JW. Nitric oxide is an early mediator of the increase in bone formation by mechanical stimulation. Am J Physiol. 1996;270:E955-60.44.
Fox SW, Chow JWM. Nitric oxide synthase expression in bone cells. Bone. 1998;23:1-6.45.
Franz‐Odendaal TA, Hall BK, Witten PE. Buried alive: How osteoblasts become osteocytes. Dev Dyn. 2006;235:176-90.46.
Gasser JA, Kneissel M. Bone physiology and biology. In: Smith S, Varela A, Samadfam R (eds): Bone toxicology: Molecular and integrative toxicology (pp 27-94). Cham: Springer, 2017.47.
Gattereau A, Bielmann P, Durivage J, Davignon J, Larochelle P. Effect of acute and chronic administration of calcitonin on serum glucose in patients with Paget's disease of bone. J Clin Endocrinol Metab. 1980;51:354-7.48.
Ghasemi A, Jeddi S. Anti-obesity and anti-diabetic effects of nitrate and nitrite. Nitric Oxide. 2017;70:9-24.49.
González‐Zabaleta J, Pita‐Fernandez S, Seoane‐Pillado T, López‐Calviño B, Gonzalez‐Zabaleta JL. Comorbidity as a predictor of mortality and mobility after hip fracture. Geriatr Gerontol Int. 2016;16:561-9.50.
Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103:137-49.51.
Gyurko R, Shoji H, Battaglino RA, Boustany G, Gibson FC 3rd, Genco CA, et al. Inducible nitric oxide synthase mediates bone development and P. gingivalis-induced alveolar bone loss. Bone. 2005;36:472-9.52.
Hagiwara H, Inoue A, Yamaguchi A, Yokose S, Furuya M, Tanaka S, et al. cGMP produced in response to ANP and CNP regulates proliferation and differentiation of osteoblastic cells. Am J Physiol Cell Physiol. 1996;270:C1311-8.53.
Hamann C, Goettsch C, Mettelsiefen J, Henkenjohann V, Rauner M, Hempel U, et al. Delayed bone regeneration and low bone mass in a rat model of insulin-resistant type 2 diabetes mellitus is due to impaired osteoblast function. Am J Physiol Endocrinol Metab. 2011;301:E1220-8.54.
Hao YJ, Tang Y, Chen FB, Pei FX. Different doses of nitric oxide donor prevent osteoporosis in ovariectomized rats. Clin Orthop Relat Res. 2005;435:226-31.55.
Hefler LA, Reyes CA, O'Brien WE, Gregg AR. Perinatal development of endothelial nitric oxide synthase-deficient mice. Biol Reprod. 2001;64:666-73.56.
Heino TJ, Hentunen TA, Väänänen HK. Osteocytes inhibit osteoclastic bone resorption through transforming growth factor-beta: Enhancement by estrogen. J Cell Biochem. 2002;85:185-97.57.
Helfrich MH, Evans DE, Grabowski PS, Pollock JS, Ohshima H, Ralston SH. Expression of nitric oxide synthase isoforms in bone and bone cell cultures. J Bone Mineral Res. 1997;12:1108-15.58.
Henriksen K, Bollerslev J, Everts V, Karsdal MA. Osteoclast activity and subtypes as a function of physiology and pathology - implications for future treatments of osteoporosis. Endocr Rev. 2011;32:31-63.59.
Hikiji H, Shin WS, Oida S, Takato T, Koizumi T, Toyo-oka T. Direct action of nitric oxide on osteoblastic differentiation. FEBS Lett. 1997;410:238-42.60.
Ho W-P, Chen T-L, Chiu W-T, Tai Y-t, Chen R-M. Nitric oxide induces osteoblast apoptosis through a mitochondria-dependent pathway. Ann N Y Acad Sci. 2005;1042:460-70.61.
Hof RJVT, Ralston SH. Nitric oxide and bone. Immunology. 2001;103:255-61.62.
Hofbauer LC, Brueck CC, Singh SK, Dobnig H. Osteoporosis in patients with diabetes mellitus. J Bone Mineral Res. 2007;22:1317-28.63.
Ho-Pham LT, Nguyen TV. Association between trabecular bone score and type 2 diabetes: A quantitative update of evidence. Osteoporos Int. 2019;30:2079-85.64.
Hukkanen M, Platts LAM, Lawes T, Girgis SI, Konttinen YT, Goodship AE, et al. Effect of nitric oxide donor nitroglycerin on bone mineral density in a rat model of estrogen deficiency-induced osteopenia. Bone. 2003;32:142-9.65.
Hukkanen MV, Platts LA, Fernandez De Marticorena I, O'Shaughnessy M, MacIntyre I, Polak JM. Developmental regulation of nitric oxide synthase expression in rat skeletal bone. J Bone Mineral Res. 1999;14:868-77.66.
Hygum K, Starup-Linde J, Harsløf T, Vestergaard P, Langdahl BL. Mechanisms in endocrinology: Diabetes mellitus, a state of low bone turnover - a systematic review and meta-analysis. Eur J Endocrinol. 2017;176:R137-57.67.
Ilich JZ, Kerstetter JE. Nutrition in bone health revisited: A story beyond calcium. J Am Coll Nutr. 2000;19:715-37.68.
Inaba M, Terada M, Koyama H, Yoshida O, Ishimura E, Kawagishi T, et al. Influence of high glucose on 1,25-dihydroxyvitamin D3-induced effect on human osteoblast-like MG-63 cells. J Bone Mineral Res. 1995;10:1050-6.69.
Inoue A, Hiruma Y, Hirose S, Yamaguchi A, Hagiwara H. Reciprocal regulation by cyclic nucleotides of the differentiation of rat osteoblast-like cells and mineralization of nodules. Biochem Biophys Res Commun. 1995;215:1104-10.70.
Jamal SA, Cummings SR, Hawker GA. Isosorbide mononitrate increases bone formation and decreases bone resorption in postmenopausal women: a randomized trial. J Bone Mineral Res. 2004;19:1512-7.71.
Jamal SA, Hamilton CJ. Nitric oxide donors for the treatment of osteoporosis. Curr Osteoporos Rep. 2012;10:86-92.72.
Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol. 2007;166:495-505.73.
Jindal M, Lakhwani O, Kaur O, Agarwal S, Garg K. Bone density versus bone quality as a predictor of bone strength. Orthop Rheumatol Open Access J. 2018;12:555830.74.
John MR, Widler L, Gamse R, Buhl T, Seuwen K, Breitenstein W, et al. ATF936, a novel oral calcilytic, increases bone mineral density in rats and transiently releases parathyroid hormone in humans. Bone. 2011;49:233-41.75.
Johnell O, Kanis J. An estimate of the worldwide prevalence, mortality and disability associated with hip fracture. Osteoporos Int. 2004;15:897-902.76.
Joshua J, Schwaerzer GK, Kalyanaraman H, Cory E, Sah RL, Li M, et al. Soluble guanylate cyclase as a novel treatment target for osteoporosis. Endocrinology. 2014;155:4720-30.77.
Jung JY, Lin AC, Ramos LM, Faddis BT, Chole RA. Nitric oxide synthase I mediates osteoclast activity in vitro and in vivo. J Cell Biochem. 2003;89:613-21.78.
Kalyanaraman H, Ramdani G, B Pilz R. Targeting NO signaling for the treatment of osteoporosis. Curr Med Chem. 2016;23:2746-53.79.
Kalyanaraman H, Schall N, Pilz RB. Nitric oxide and cyclic GMP functions in bone. Nitric oxide. 2018;76:62-70.80.
Kalyanaraman H, Schwaerzer G, Ramdani G, Castillo F, Scott BT, Dillmann W, et al. Protein kinase G activation reverses oxidative stress and restores osteoblast function and bone formation in male mice with type 1 diabetes. Diabetes. 2018;67:607-23.81.
Kalyanaraman H, Schwappacher R, Joshua J, Zhuang S, Scott BT, Klos M, et al. Nongenomic thyroid hormone signaling occurs through a plasma membrane–localized receptor. Sci Signal. 2014;7 (326):ra48.82.
Kanazawa I. Interaction between bone and glucose metabolism [Review]. Endocr J. 2017;64:1043-53.83.
Kapil V, Khambata RS, Jones DA, Rathod K, Primus C, Massimo G, et al. The noncanonical pathway for in vivo nitric oxide generation: The nitrate-nitrite-nitric oxide pathway. Pharmacol Rev. 2020;72:692-766.84.
Karras SN, Anagnostis P, Antonopoulou V, Tsekmekidou X, Koufakis T, Goulis DG, et al. The combined effect of vitamin D and parathyroid hormone concentrations on glucose homeostasis in older patients with prediabetes: A cross-sectional study. Diab Vasc Dis Res. 2018;15:150-3.85.
Karst M, Gorny G, Galvin RJ, Oursler MJ. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-beta regulation of osteoclast differentiation. J Cell Physiol. 2004;200:99-106.86.
Kasten TP, Collin-Osdoby P, Patel N, Osdoby P, Krukowski M, Misko TP, et al. Potentiation of osteoclast bone-resorption activity by inhibition of nitric oxide synthase. Proc Natl Acad Sci U S A. 1994;91:3569-73.87.
Khosla S, Burr D, Cauley J, Dempster DW, Ebeling PR, Felsenberg D, et al. Bisphosphonate-associated osteonecrosis of the jaw: Report of a task force of the American Society for Bone and Mineral Research. J Bone Mineral Res. 2007;22:1479-91.88.
Khosla S, Hofbauer LC. Osteoporosis treatment: Recent developments and ongoing challenges. The Lancet Diabetes Endocrinol. 2017;5:898-907.89.
Klein-Nulend J, Helfrich MH, Sterck JGH, MacPherson H, Joldersma M, Ralston SH, et al. Nitric oxide response to shear stress by human bone cell cultures is endothelial nitric oxide synthase dependent. Biochem Biophys Res Communs. 1998;250:108-14.90.
Lattanzio S, Santilli F, Liani R, Vazzana N, Ueland T, Di Fulvio P, et al. Circulating dickkopf-1 in diabetes mellitus: association with platelet activation and effects of improved metabolic control and low-dose aspirin. J Am Heart Assoc. 2014;3:e001000.91.
Li K, Wang X-F, Li D-Y, Chen Y-C, Zhao L-J, Liu X-G, et al. The good, the bad, and the ugly of calcium supplementation: a review of calcium intake on human health. Clin Interv Aging. 2018;13:2443-52.92.
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883-7.93.
Lim SY, Bolster MB. Profile of romosozumab and its potential in the management of osteoporosis. Drug Des Devel Ther. 2017;11:1221-31.94.
Lipscombe LL, Jamal SA, Booth GL, Hawker GA. The risk of hip fractures in older individuals with diabetes: A population-based study. Diabetes Care. 2007;30:835-41.95.
Lundberg JO, Carlström M, Weitzberg E. Metabolic effects of dietary nitrate in health and disease. Cell Metab. 2018;28:9-22.96.
MacPherson H, Noble BS, Ralston SH. Expression and functional role of nitric oxide synthase isoforms in human osteoblast-like cells. Bone. 1999;24:179-85.97.
Mancini L, Moradi-Bidhendi N, Becherini L, Martineti V, MacIntyre I. The biphasic effects of nitric oxide in primary rat osteoblasts are cGMP dependent. Biochem Biophys Res Commun. 2000;274:477-81.98.
Mathieu C, Gysemans C, Giulietti A, Bouillon R. Vitamin D and diabetes. Diabetologia. 2005;48:1247-57.99.
Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. 2013;34:309-38.100.
McFarlane SI, Muniyappa R, Shin JJ, Bahtiyar G, Sowers JR. Osteoporosis and cardiovascular disease. Endocrine. 2004;23:1-10.101.
Moayeri A, Mohamadpour M, Mousavi SF, Shirzadpour E, Mohamadpour S, Amraei M. Fracture risk in patients with type 2 diabetes mellitus and possible risk factors: S systematic review and meta-analysis. Ther Clin Risk Manag. 2017;13:455-68.102.
Mohsin S, Baniyas MMYH, AlDarmaki RSMH, Tekes K, Kalász H, Adeghate EA. An update on therapies for the treatment of diabetes-induced osteoporosis. Expert Opin Biol Ther. 2019;19:937-48.103.
Mordwinkin NM, Meeks CJ, Jadhav SS, Espinoza T, Roda N, diZerega GS, et al. Angiotensin-(1-7) administration reduces oxidative stress in diabetic bone marrow. Endocrinology. 2012;153:2189-97.104.
Morita M, Sato Y, Iwasaki R, Kobayashi T, Watanabe R, Oike T, et al. Selective estrogen receptor modulators suppress hif1α protein accumulation in mouse osteoclasts. PLoS One. 2016;11:e0165922.105.
Mundy GR. Cytokines and growth factors in the regulation of bone remodeling. J Bone Mineral Res. 1993;8 (Suppl 2):S505-10.106.
Münzel T, Daiber A. Inorganic nitrite and nitrate in cardiovascular therapy: A better alternative to organic nitrates as nitric oxide donors? Vasc Pharmacol. 2018;102:1-10.107.
Nabhan AF, Rabie NH. Isosorbide mononitrate versus alendronate for postmenopausal osteoporosis. Int J Gynaecol Obstet. 2008;103:213-6.108.
Napoli N, Strollo R, Defeudis G, Leto G, Moretti C, Zampetti S, et al. Serum sclerostin and bone turnover in latent autoimmune diabetes in adults. J Clin Endocrinol Metab. 2018;103:1921-8.109.
Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434-41.110.
Nemeth EF, Delmar EG, Heaton WL, Miller MA, Lambert LD, Conklin RL, et al. Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J Pharmacol Exp Ther. 2001;299:323-31.111.
Nemeth EF, Goodman WG. Calcimimetic and calcilytic drugs: Feats, flops, and futures. Calcif Tissue Int. 2016;98:341-58.112.
Norris R, Parker M. Diabetes mellitus and hip fracture: A study of 5966 cases. Injury. 2011;42:1313-6.113.
Oei L, Rivadeneira F, Zillikens MC, Oei EHG. Diabetes, diabetic complications, and fracture risk. Curr Osteoporos Rep. 2015;13:106-15.114.
Palermo A, D'Onofrio L, Buzzetti R, Manfrini S, Napoli N. Pathophysiology of bone fragility in patients with diabetes. Calcif Tissue Int. 2017;100:122-32.115.
Patsch JM, Burghardt AJ, Yap SP, Baum T, Schwartz AV, Joseph GB, et al. Increased cortical porosity in type 2 diabetic postmenopausal women with fragility fractures. J Bone Mineral Re. 2013;28:313-24.116.
Percival MD, Ouellet M, Campagnolo C, Claveau D, Li C. Inhibition of cathepsin K by nitric oxide donors: evidence for the formation of mixed disulfides and a sulfenic acid. Biochemistry. 1999;38:13574-83.117.
Perez-Diaz I, Sebastian-Barajas G, Hernandez-Flores ZG, Rivera-Moscoso R, Osorio-Landa HK, Flores-Rebollar A. The impact of vitamin D levels on glycemic control and bone mineral density in postmenopausal women with type 2 diabetes. J Endocrinol Invest. 2015;38:1365-72.118.
Pettway GJ, Meganck JA, Koh AJ, Keller ET, Goldstein SA, McCauley LK. Parathyroid hormone mediates bone growth through the regulation of osteoblast proliferation and differentiation. Bone. 2008;42:806-18.119.
Pezhman L, Sheikhzadeh Hesari F, Ghiasi R, Alipour MR. The impact of forced swimming on expression of RANKL and OPG in a type 2 diabetes mellitus rat model. Arch Physiol Biochem. 2019;125:195-200.120.
Picke AK, Campbell G, Napoli N, Hofbauer LC, Rauner M. Update on the impact of type 2 diabetes mellitus on bone metabolism and material properties. Endocr Connect. 2019;8:R55-70.121.
Picke AK, Salbach-Hirsch J, Hintze V, Rother S, Rauner M, Kascholke C, et al. Sulfated hyaluronan improves bone regeneration of diabetic rats by binding sclerostin and enhancing osteoblast function. Biomaterials. 2016;96:11-23.122.
Pouwels S, Lalmohamed A, van Staa T, Cooper C, Souverein P, Leufkens HG, et al. Use of organic nitrates and the risk of hip fracture: a population-based case-control study. J Clin Endocrinol Metab. 2010;95:1924-31.123.
Pritchard JM, Papaioannou A, Tomowich C, Giangregorio LM, Atkinson SA, Beattie KA, et al. Bone mineralization is elevated and less heterogeneous in adults with type 2 diabetes and osteoarthritis compared to controls with osteoarthritis alone. Bone. 2013;54:76-82.124.
Pun KK, Lau P, Ho PWM. The characterization, regulation, and function of insulin receptors on osteoblast-like clonal osteosarcoma cell line. J Bone Mineral Res. 1989;4:853-62.125.
Punjabi CJ, Laskin DL, Heck DE, Laskin JD. Production of nitric oxide by murine bone marrow cells. Inverse correlation with cellular proliferation. J Immunol. 1992;149:2179-84.126.
Purnamasari D, Puspitasari MD, Setiyohadi B, Nugroho P, Isbagio H. Low bone turnover in premenopausal women with type 2 diabetes mellitus as an early process of diabetes-associated bone alterations: a cross-sectional study. BMC Endocr Disord. 2017;17:72.127.
Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem. 2010;285:25103-8.128.
Rajfer RA, Flores M, Abraham A, Garcia E, Hinojosa N, Desai M, et al. Prevention of osteoporosis in the ovariectomized rat by oral administration of a nutraceutical combination that stimulates nitric oxide production. J Osteoporos. 2019;2019:1592328.129.
Ralston SH, Ho LP, Helfrich MH, Grabowski PS, Johnston PW, Benjamin N. Nitric oxide: A cytokine-induced regulator of bone resorption. J Bone Mineral Res. 1995;10:1040-9.130.
Ralston SH, Todd D, Helfrich M, Benjamin N, Grabowski PS. Human osteoblast-like cells produce nitric oxide and express inducible nitric oxide synthase. Endocrinology. 1994;135:330-6.131.
Rejnmark L, Vestergaard P, Mosekilde L. Decreased fracture risk in users of organic nitrates: a nationwide case-control study. J Bone Mineral Res. 2006;21:1811-7.132.
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866-75.133.
Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, et al. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611-7.134.
Russell RGG. Bisphosphonates: The first 40 years. Bone. 2011;49:2-19.135.
Russo J, Russo IH. The role of estrogen in the initiation of breast cancer. J Steroid Biochem Mol Biol. 2006;102:89-96.136.
Samuels A, Perry MJ, Gibson RL, Colley S, Tobias JH. Role of endothelial nitric oxide synthase in estrogen-induced osteogenesis. Bone. 2001;29:24-9.137.
Sarkar PD, Choudhury AB. Relationships between serum osteocalcin levels versus blood glucose, insulin resistance and markers of systemic inflammation in central Indian type 2 diabetic patients. Eur Rev Med Pharmacol Sci. 2013;17:1631-5.138.
Saura M, Tarin C, Zaragoza C. Recent insights into the implication of nitric oxide in osteoblast differentiation and proliferation during bone development. ScientificWorldJournal. 2010;10:624-32.139.
Schmidt HH, Gagne GD, Nakane M, Pollock JS, Miller MF, Murad F. Mapping of neural nitric oxide synthase in the rat suggests frequent co-localization with NADPH diaphorase but not with soluble guanylyl cyclase, and novel paraneural functions for nitrinergic signal transduction. J Histochem Cytochem. 1992;40:1439-56.140.
Sosa M, Dominguez M, Navarro MC, Segarra MC, Hernandez D, de Pablos P, et al. Bone mineral metabolism is normal in non-insulin-dependent diabetes mellitus. J Diabetes Complications. 1996;10:201-5.141.
Stabley JN, Prisby RD, Behnke BJ, Delp MD. Type 2 diabetes alters bone and marrow blood flow and vascular control mechanisms in the ZDF rat. J Endocrinol. 2015;225:47-58.142.
Tai YT, Cherng YG, Chang CC, Hwang YP, Chen JT, Chen RM. Pretreatment with low nitric oxide protects osteoblasts from high nitric oxide-induced apoptotic insults through regulation of c-Jun N-terminal kinase/c-Jun-mediated Bcl-2 gene expression and protein translocation. J Orthop Res. 2007;25:625-35.143.
Takakura A, Lee J-W, Hirano K, Isogai Y, Ishizuya T, Takao-Kawabata R, et al. Administration frequency as well as dosage of PTH are associated with development of cortical porosity in ovariectomized rats. Bone Res. 2017;5:17002.144.
Tella SH, Gallagher JC. Biological agents in management of osteoporosis. Eur J Clin Pharmacol. 2014;70:1291-301.145.
Tella SH, Kommalapati A, Correa R. Profile of abaloparatide and its potential in the treatment of postmenopausal osteoporosis. Cureus. 2017;9:e1300.146.
Thum T, Fraccarollo D, Schultheiss M, Froese S, Galuppo P, Widder JD, et al. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes. 2007;56:666-74.147.
Tsourdi E, Langdahl B, Cohen-Solal M, Aubry-Rozier B, Eriksen EF, Guañabens N, et al. Discontinuation of Denosumab therapy for osteoporosis: A systematic review and position statement by ECTS. Bone. 2017;105:11-7.148.
van Bezooijen RL, Roelen BAJ, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805-14.149.
van Daele PLA, Stolk RP, Burger H, Algra D, Grobbee DE, Hofman A, et al. Bone density in non-insulin-dependent diabetes mellitus: The rotterdam study. Ann Intern Med. 1995;122:409-14.150.
van’t Hof RJ, MacPhee J, Libouban H, Helfrich MH, Ralston SH. Regulation of bone mass and bone turnover by neuronal nitric oxide synthase. Endocrinology. 2004;145:5068-5074.151.
Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—a meta-analysis. Osteoporos Int. 2007;18:427-44.152.
Wang C-B, Lin C-FJ, Liang W-M, Cheng C-F, Chang Y-J, Wu H-C, et al. Excess mortality after hip fracture among the elderly in Taiwan: a nationwide population-based cohort study. Bone. 2013;56:147-53.153.
Ward LM, Rauch F. Anabolic therapy for the treatment of osteoporosis in childhood. Curr Osteoporos Rep. 2018;16:269-76.154.
Watanuki M, Sakai A, Sakata T, Tsurukami H, Miwa M, Uchida Y, et al. Role of inducible nitric oxide synthase in skeletal adaptation to acute increases in mechanical loading. J Bone Mineral Res. 2002;17:1015-25.155.
Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H, et al. Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell. 2015;161:1576-91.156.
Wimalawansa S. Nitric oxide: Novel therapy for osteoporosis. Expert Opin Pharmacother. 2009;9:3025-44.157.
Wimalawansa S, Chapa T, Fang L, Yallampalli C, Simmons D, Wimalawansa S. Frequency-dependent effect of nitric oxide donor nitroglycerin on bone. J Bone Mineral Res. 2000;15:1119-25.158.
Wimalawansa SJ. Nitric oxide and bone. Ann N Y Acad Sci. 2010;1192:391-403.159.
Wimalawansa SJ. Nitroglycerin therapy is as efficacious as standard estrogen replacement therapy (Premarin) in prevention of oophorectomy-induced bone loss: a human pilot clinical study. J Bone Mineral Res. 2000;15:2240-4.160.
Wimalawansa SJ. Rationale for using nitric oxide donor therapy for prevention of bone loss and treatment of osteoporosis in humans. Ann N Y Acad Sci. 2007;1117:283-97.161.
Wimalawansa SJ. Restoration of ovariectomy-induced osteopenia by nitroglycerin. Calcif Tissue Int. 2000;66:56-60.162.
Wimalawansa SJ. Skeletal effects of nitric oxide novel agent for osteoporosis. In: Bilezikian JP, Raisz LG, John Martin T (eds): Principles of bone biology, 3rd ed. (pp 1273-310). Amsterdam: Elsevier, 2008.163.
Wimalawansa SJ, De Marco G, Gangula P, Yallampalli C. Nitric oxide donor alleviates ovariectomy-induced bone loss. Bone. 1996;18:301-4.164.
Wongdee K, Charoenphandhu N. Osteoporosis in diabetes mellitus: Possible cellular and molecular mechanisms. World J Diabetes. 2011;2:41-8.165.
Yan Q, Feng Q, Beier F. Endothelial nitric oxide synthase deficiency in mice results in reduced chondrocyte proliferation and endochondral bone growth. Arthritis Rheum. 2010;62:2013-22.166.
Yang S, Guo L, Su Y, Wen J, Du J, Li X, et al. Nitric oxide balances osteoblast and adipocyte lineage differentiation via the JNK/MAPK signaling pathway in periodontal ligament stem cells. Stem Cell Res Ther. 2018;9:118.167.
Youn JY, Zhou J, Cai H. Bone morphogenic protein 4 mediates NOX1-dependent eNOS uncoupling, endothelial dysfunction, and COX2 induction in type 2 diabetes mellitus. Mol Endocrinol. 2015;29:1123-33.168.
Zaragoza C, Lopez-Rivera E, Garcia-Rama C, Saura M, Martinez-Ruiz A, Lizarbe TR, et al. Cbfa-1 mediates nitric oxide regulation of MMP-13 in osteoblasts. J Cell Sci. 2006;119:1896-902.169.
Zhang N, Zhang ZK, Yu Y, Zhuo Z, Zhang G, Zhang BT. Pros and cons of denosumab treatment for osteoporosis and implication for RANKL aptamer therapy. Front Cell Dev Biol. 2020;8:325.170.
Zhao S, Mugabo Y, Ballentine G, Attane C, Iglesias J, Poursharifi P, et al. α/β-hydrolase domain 6 deletion induces adipose browning and prevents obesity and type 2 diabetes. Cell Rep. 2016;14:2872-88.171.
Zheng H, Yu X, Collin-Osdoby P, Osdoby P. RANKL stimulates inducible nitric-oxide synthase expression and nitric oxide production in developing osteoclasts. An autocrine negative feedback mechanism triggered by RANKL-induced interferon-beta via NF-kappaB that restrains osteoclastogenesis and bone resorption. J Biol Chem. 2006;281:15809-20.172.
Zhu W, Diwan AD, Lin JH, Murrell GA. Nitric oxide synthase isoforms during fracture healing. J Bone Mineral Res. 2001;16:535-40.
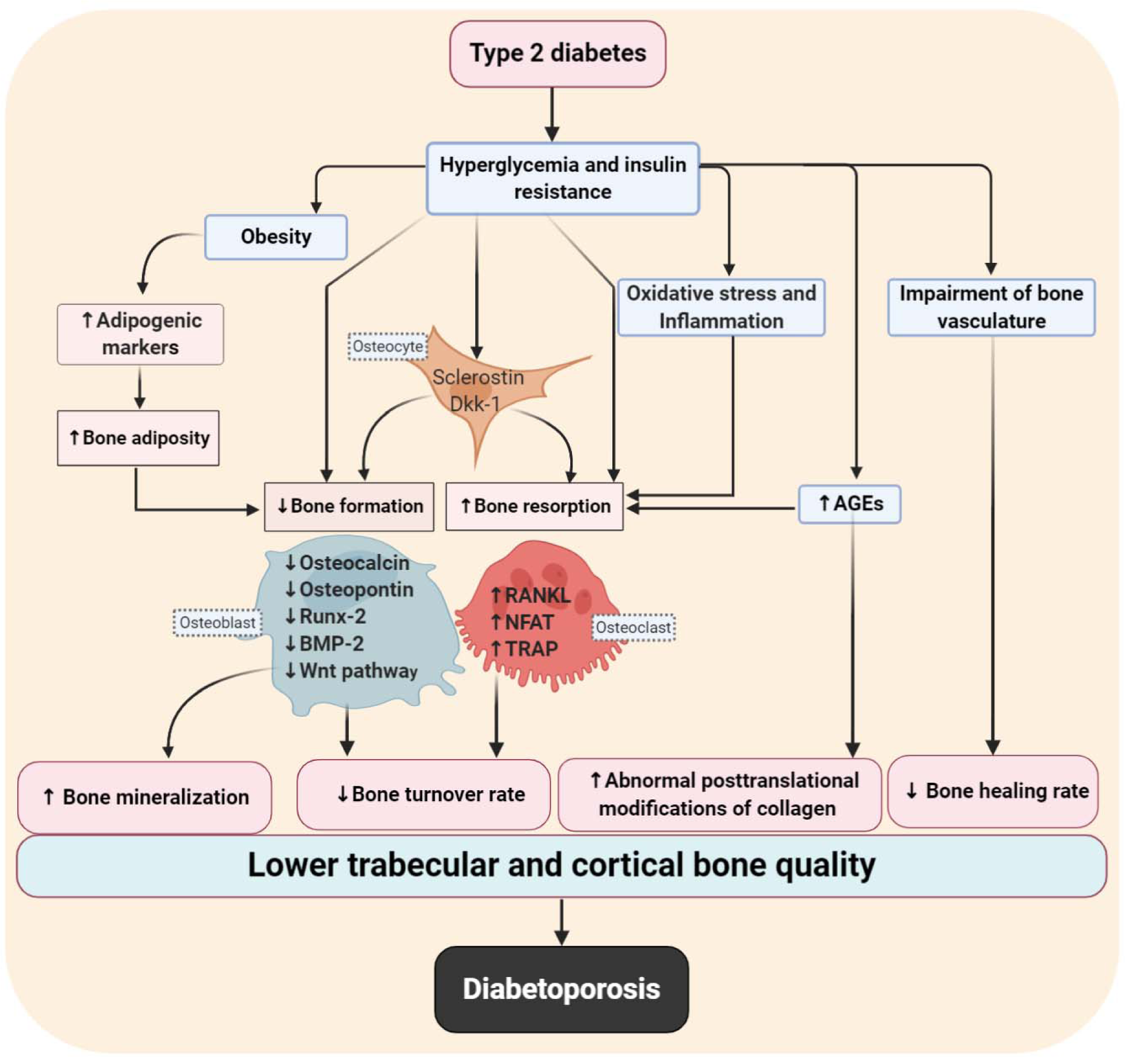
Figure 1: Main pathophysiological mechanisms involved in diabetoporosis. Type 2 diabetes (T2D) decreases trabecular and cortical bone quality by decreasing bone turnover and healing rates and increasing bone mineralization and abnormal posttranslational modifications of collagen.
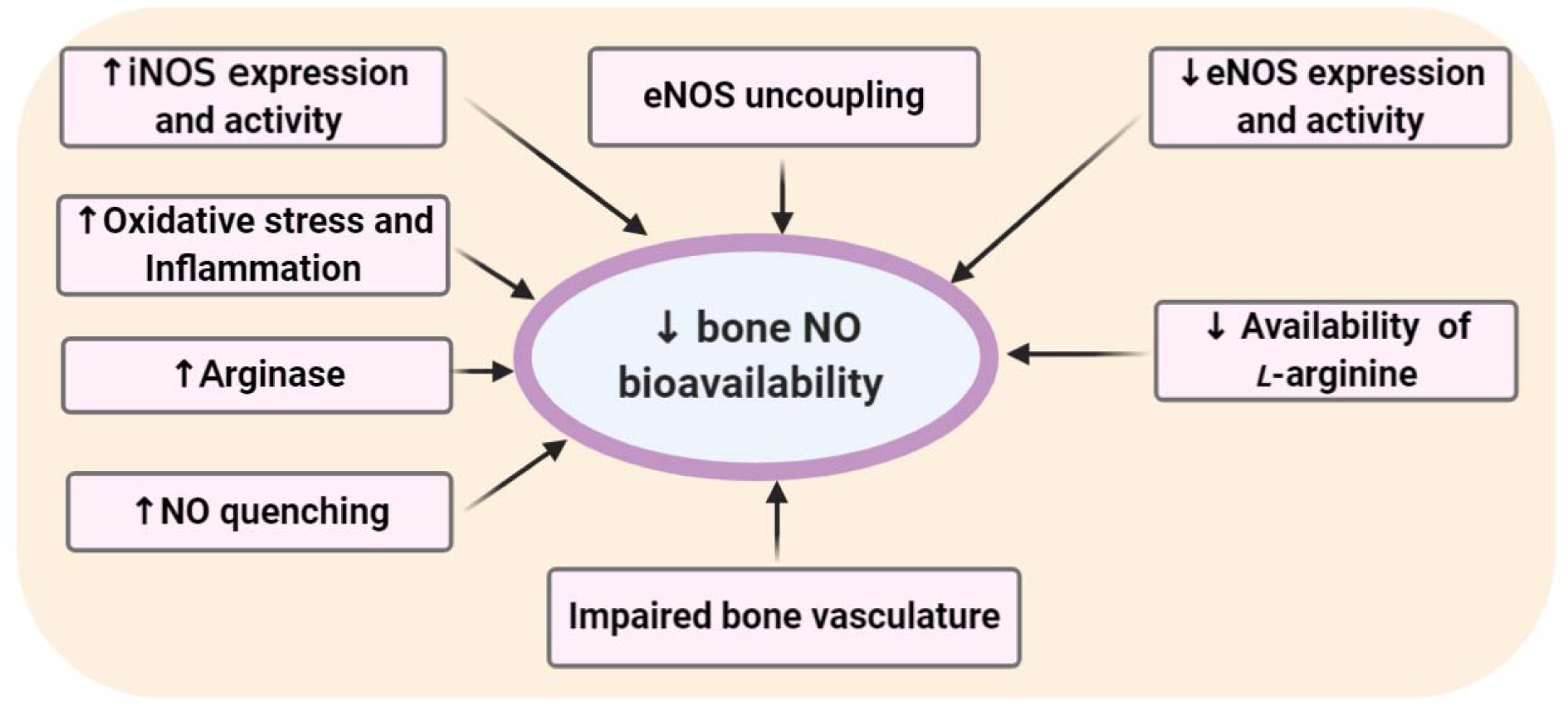
Figure 2: Proposed mechanisms involved in decreased endothelial nitric oxide (eNOS)-derived NO bioavailability and activity in bones of type 2 diabetic subjects
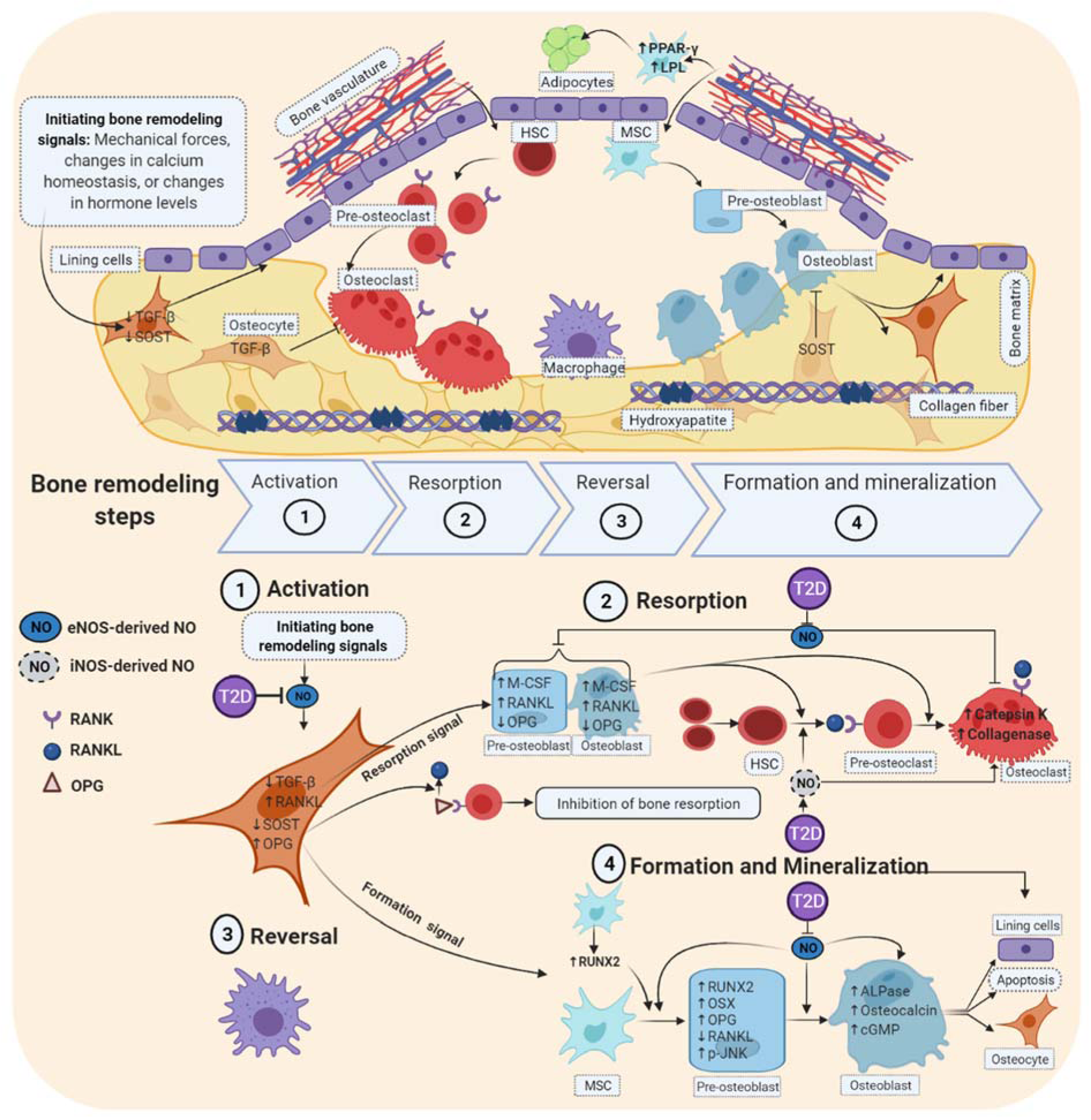
Figure 3: Effects of type 2 diabetes on bone remodeling: role of nitric oxide. T2D decreases osteoblastic bone formation and has a stimulatory effect on osteoclast-mediated bone resorption. These effects are mediated in part by a decrease in eNOS-derived NO and an increase in iNOS-derived NO.
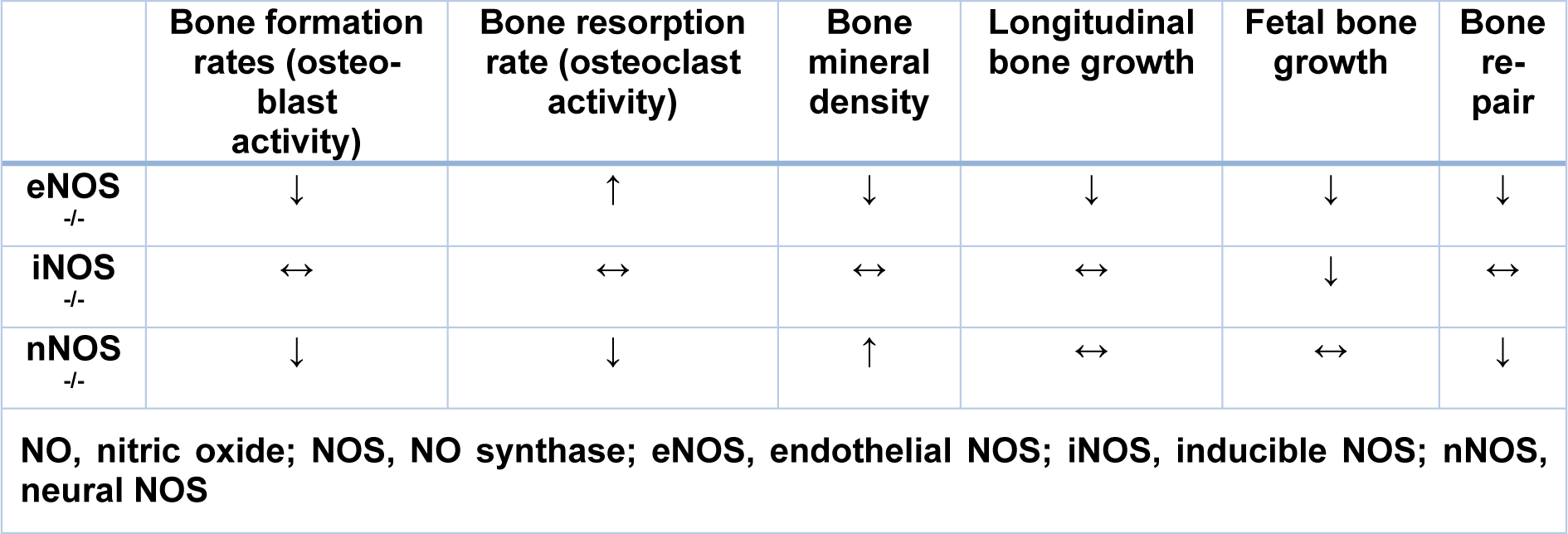
Table 1: The bone phenotypes in NOS-deficient rats and mice (Diwan et al., 2000; Armour et al., 2001; Hefler et al., 2001; Watanuki et al., 2002; Jung et al., 2003; van't Hof et al., 2004; Yan et al., 2010)
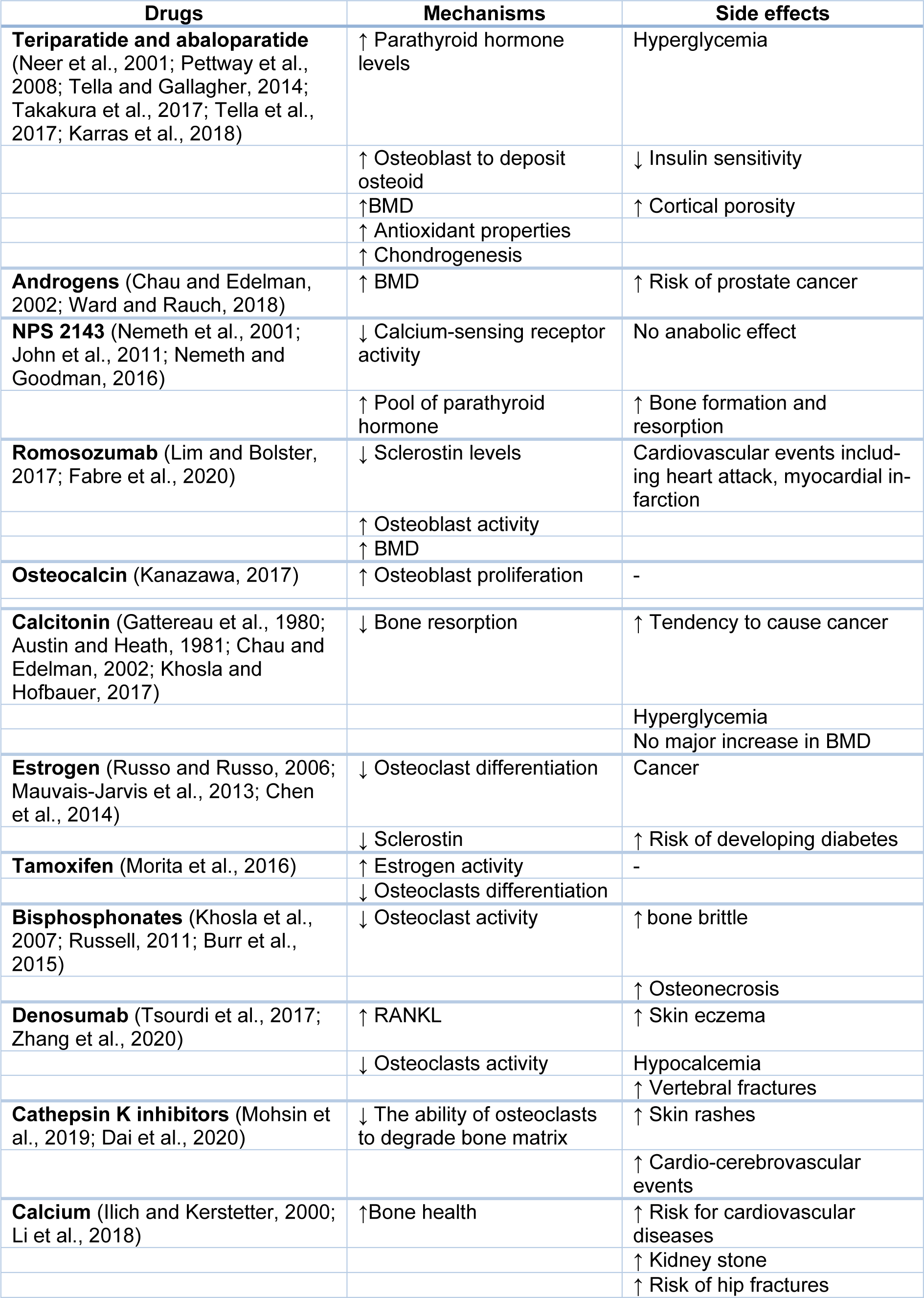
Table 2: Mechanisms and side effects of current drugs used in treating of osteoporosis