
Cardiac amyloidosis
evolving pathogenesis, multimodal diagnostics, and principles of treatment
DOI:
https://doi.org/10.17179/excli2023-6284Keywords:
cardiac amyloidosis, ATTR amyloidosis, gene therapy, amyloid, cardiomyopathy, CRISPRAbstract
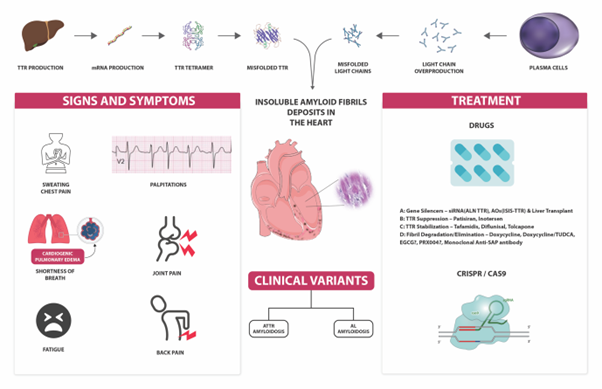
Amyloidosis is a protein deposition disorder in which insoluble fibril structures accumulate in the bodily tissues damaging the organ function. Cardiac amyloidosis is a severe but under-reported medical condition characterized by the accumulation of amyloid in the extracellular area of the myocardium, which results in thickening and stiffening of ventricular walls. Cardiac amyloidosis has recently gained much attention with its slowly surging incidence. With this study, we seek to comprehensively compile the pathophysiology and clinical picture of cardiac amyloidosis subtypes, extending a clinically oriented, up-to-date clinical approach to diagnosis and therapy. Cardiac amyloidosis can be caused by rare genetic mutations which may be inherited or acquired. The growing incidence can be attributed to advancements in imaging methods and other diagnostic modalities. Most occurrences of cardiac amyloidosis result from two forms of precursor protein: transthyretin [TTR] amyloid and immunoglobulin-derived light-chain amyloid. Prompt identification of cardiac amyloidosis can facilitate the implementation of evolving therapeutic interventions to enhance the outcomes. The modalities for the management of CA have evolved significantly in the last ten years. Apart from therapies for modifying disease and heart failure, a myriad of novel therapeutic approaches that target specific aspects of the disease, including gene therapies, are being researched. These aim at impeding its progression and improving clinical outcomes.
Downloads
Published
How to Cite
License
Copyright (c) 2023 Gnana Deepthi Medarametla, Ripudaman Singh Kahlon, Lampimukhi Mahitha, Sanobar Shariff, Naga Praneeth Vakkalagadda, Hitesh Chopra, Mohammad Amjad Kamal, Neil Patel, Yashendra Sethi, Nirja Kaka

This work is licensed under a Creative Commons Attribution 4.0 International License.
Authors who publish in this journal agree to the following terms:
- The authors keep the copyright and grant the journal the right of first publication under the terms of the Creative Commons Attribution license, CC BY 4.0. This licencse permits unrestricted use, distribution and reproduction in any medium, provided that the original work is properly cited.
- The use of general descriptive names, trade names, trademarks, and so forth in this publication, even if not specifically identified, does not imply that these names are not protected by the relevant laws and regulations.
- Because the advice and information in this journal are believed to be true and accurate at the time of publication, neither the authors, the editors, nor the publisher accept any legal responsibility for any errors or omissions presented in the publication. The publisher makes no guarantee, express or implied, with respect to the material contained herein.
- The authors can enter into additional contracts for the non-exclusive distribution of the journal's published version by citing the initial publication in this journal (e.g. publishing in an institutional repository or in a book).


